 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Revista Colombia Médica, Vol. 35, No. 1, 2004, pp. 5-11 Biodisponibilidad comparativa entre dos formulaciones de gabapentina cápsulas de 300 mg en voluntarios sanos colombianos1 Sergio Parra, M.D., M.Sc.2, Fanny Cuesta, Ing. Q. Esp.2, Margarita Restrepo, Q.F., M.Sc.3, Rosendo Archbold, Q.F., M.Sc.3, Blanca Montoya, Bact.4, Gloria Holguín, Q.F. Esp.3, Juan Carlos Ríos, M.D.5
Recibido para publicación
septiembre 18, 2003 Code Number: rc04002 RESUMEN Introducción: Comparar la
biodisponibilidad entre dos productos equivalentes
farmacéuticos, permite declarar la bioequivalencia o no de dos
medicamentos sometidos a estudio bajo condiciones experimentales
similares. Palabras claves: Gabapentina. Biodisponibilidad. Bioequivalencia. SUMMARY Introduction: To compare the
bioavailability between two pharmaceutical equivalent products. To
allow the demonstration of the bioequivalence or not, of two
formulations submitted to study under similar experimental
conditions. Key words: Gabapentin. Bioavailability. Bioequivalence. La gabapentina, ácido 1-(aminometil-1-ciclohexil) acético, es un agente antiepiléptico con propiedades de un aminoácido, soluble en agua y aprobado para ser vendido en los Estados Unidos de América desde 19931,2. Aunque esta molécula está estructuralmente relacionada con el neurotransmisor GABA (ácido gamma-amino butírico), no interactúa como agonista con los receptores GABA, no es convertido metabólicamente en GABA y no es inhibidor de la recaptación del GABA o degradación3, pero eleva la liberación del GABA desde las células gliales por un mecanismo molecular aún no esclarecido4. La gabapentina circula, en su mayoría, sin unirse a las proteínas plasmáticas; la concentración plasmática máxima se alcanza entre las 2 y 3 horas, con farmacocinética lineal, con un volumen de distribución de 57.7 l/kg y es eliminada de la circulación sistémica por excreción renal como sustancia sin metabolizar. La vida media de eliminación es de 5 a 7 horas. La constante de velocidad de eliminación, la depuración plasmática y la depuración renal son directamente proporcionales a la depuración de la creatinina5. La biodisponibilidad de la gabapentina es cercana a 60%, no es proporcional a la dosis, sin embargo las diferencias en biodisponibilidad no son grandes. Los alimentos no tienen efecto sobre la velocidad y extensión de la absorción. Este medicamento está indicado como terapia de adición en la epilepsia, para crisis parciales con o sin generalización secundaria, en pacientes que no han alcanzado un control satisfactorio o no han tolerado los anticonvulsivantes de primera línea y en los síndromes de dolor neuropático6. Como es un medicamento de amplia prescripción, se hace necesario tener otras fuentes de adquisición, pero sin detrimento de su calidad, por tanto es un deber de quien se interese en presentar otra formulación del mismo principio activo e igual forma de presentación, garantizar la calidad integral, que debe incluir la calidad biofarmacéutica que se demuestra si la velocidad y cantidad absorbida, evaluadas por las variables farmacocinéticas adecuadas, no presentan diferencias significativas con respecto a la velocidad de absorción y cantidad absorbida del producto utilizado como referencia, teniendo como base un número de voluntarios suficiente que permita garantizar el poder estadístico de la prueba. Esto es, estar seguros de que al administrarse un medicamento sólido por vía oral llegue a la circulación sistémica, en la cantidad y velocidad adecuadas para obtener el efecto terapéutico deseado. Además, si dentro de las pretensiones del laboratorio solicitante está la de obtener un certificado de intercambiabilidad también requiere del estudio de bioequivalencia para asegurarle al prescriptor que el paciente no tendrá ningún tipo de trastorno al cambiar el producto innovador por el genérico o viceversa. Para realizar los estudios de bioequivalencia en un preparado farmacéutico, este debe cumplir con dos condiciones previas:
Por lo anterior, en Colombia desde 1995, el Decreto 677 del Ministerio de Salud7 estableció la obligatoriedad de presentar con la solicitud del registro sanitario para medicamentos, los resultados de los estudios de biodisponibilidad y bioequivalencia para los productos definidos por el Instituto de Vigilancia y Control de Medicamentos y Alimentos (INVIMA); y en agosto de 2001 en la Resolución 1400, artículo 5, el Ministerio de Salud exige estudios de bioequivalencia in vivo para los anticonvulsivantes, grupo en el cual está clasificado farmacológicamente la gabapentina8. En el presente estudio para declarar la bioequivalencia se comprobó la equivalencia farmacéutica y se compararon los parámetros farmacocinéticos obtenidos a partir de los perfiles de concentración plasmática vs. tiempo, en voluntarios sanos, del producto de prueba Gabantex®/Gabapentin MK elaborado y comercializado por Tecnoquímicas S.A. en Colombia, con referencia el producto innovador Neurontin® de Parke-Davis & Company. MÉTODOS Materiales comunes
Equipos y materiales para la equivalencia farmacéutica. Disolutor Hanson Research, cromatógrafo líquido Perkin Elmer, Series 200, con identificador ultravioleta con arreglo de diodos, columna cromatográfica C18 Hewlett Packard®, controlado con el software TurboChrom®. Reactivos grado HPLC. Agua obtenida en un equipo Milli Q Plus®, acetonitrilo y metanol Merck. Reactivos grado analítico marca JT Baker. Fosfato de sodio, fosfato de potasio, ácido fosfórico, fosfato de amonio, ácido clorhídrico, hidróxido de sodio, sal sódica del ácido decanosulfónico y trietilamina. Equipos y materiales para determinar gabapentina en plasma. Cromatógrafo líquido Hewlett Packard, modelo 1100, con detector de fluorescencia, columna cromatográfica C18 LiChosorb RP-select B Merck®, controlado con el programa ChemStation®, con el cual se realizó la integración y la cuantificación del fármaco administrado. Reactivos grado HPLC. Metanol, acetonitrilo y agua. Reactivos grado analítico. Acido perclórico, ácido o-phtaldehído-3-mercaptopropiónico y EDTA. Método para la equivalencia farmacéutica. Los dos productos fueron evaluados in vitro, con el fin de verificar el cumplimiento de las especificaciones establecida por la Farmacopea Americana USP 26-NF 21 para cápsulas9, utilizando los métodos de cuantificación de gabapentina establecidos por Tecnoquímicas S.A., para las pruebas de uniformidad de dosis, disolución y valoración (contenido de gabapentina por cápsula). Criterio de aceptación. En la valoración, el producto de prueba no debe diferir en más de 5% con respecto al producto de referencia. Método para el estudio de bioequivalencia. Diseño del estudio in vivo. El estudio de bioequivalencia se realizó en 14 voluntarios sanos, 7 hombres y 7 mujeres, bajo un diseño aleatorio, cruzado, con dosis única, en dos períodos, con dos tratamientos, dos secuencias y un tiempo de lavado de una semana. Previo a la selección de los voluntarios, se presentó el protocolo del estudio al Comité de Ética de la Facultad de Medicina de la Universidad de Antioquia y al Comité de Ética del Hospital Universitario San Vicente de Paúl (HSVP) de Medellín para su aprobación. Todas las etapas del estudio in vivo se desarrollaron cumpliendo con las BPC10, los principios éticos de la Declaración de Helsinki11 actualizada en octubre de 2000 y la Resolución Nº 008430/93 del Ministerio de Salud de la República de Colombia12. Los sujetos incluidos en el estudio tuvieron edades comprendidas entre 17 y 28 años de edad, peso corporal entre 50.1 y 72.9 kg y la estatura varió en un rango de 1.55 a 1.84 metros. Todos cumplieron con las características de talla y peso establecidas por la Metropolitan Life. Estos voluntarios se sometieron a evaluación médica y exámenes paraclínicos, incluyendo VIH, hepatitis B y embarazo en las mujeres. Recibieron toda la información concerniente al estudio, los riesgos y los beneficios y las condiciones a cumplir durante el mismo. Todos firmaron el consentimiento informado antes de iniciar el estudio. El primer día de cada período, los sujetos fueron confinados en el HSVP por un día, previo ayuno de 12 horas. Antes de la administración del medicamento se tomaron muestras de orina para verificar ausencia de drogas de abuso (alcohol etílico, cocaína, marihuana, morfina, benzodiazepinas y anfetaminas); se tomaron los signos vitales y la muestra de sangre correspondiente al tiempo cero (0:0 hora). Acto seguido se les administraron 2 cápsulas (600 mg de gabapentina) del producto de prueba o del producto de referencia, de acuerdo con la tabla de asignación al azar, con 240 ml de agua. Se tomaron muestras de sangre en tubos heparinizados, a las 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 5.0, 6.0, 12.0, 24.0 y 36.0 horas. La alimentación suministrada fue estándar: desayuno, refrigerio, almuerzo, refrigerio y cena, a las 2.0, 3.5, 5.0, 8.0 y 11.0 horas respectivamente después de administrado el medicamento. Las muestras sanguíneas fueron sometidas a centrifugación para separar el plasma y dejarlo en congelación a -20ºC, hasta el momento del análisis. Análisis para los niveles plasmáticos de gabapentina. Los niveles plasmáticos de gabapentina fueron analizados usando el método de cromatografía líquida informado por Forrest et al.13. Este método fue previamente validado en un rango entre 0.0781 mg/ml y 5.0 mg/ml, con los siguientes resultados: el porcentaje promedio de recuperación fue 87.1%, mientras que el coeficiente de variación (CV) de las pendientes de las curvas de calibración fue de 4.6% para la validación intradía y 8.8% para la validación interdía; para una concentración de 0.3125 mg/ml, la precisión tuvo un CV de 11.8% y para la concentración de 5 mg/ml, el CV fue de 7.5%. Las muestras se trataron con ácido perclórico 2 M, centrifugadas y luego derivatizadas con el ácido o-phtaldehído-3-mercaptopropionico. La separación se efectuó en una columna C18, utilizando como fase móvil buffer de acetato pH 3.7-metanol-acetonitrilo (40:30:30), empleando un detector de fluorescencia programado con longitudes de onda de excitación y emisión de 330 y 440 nm respectivamente. Análisis farmacocinético. Para cada voluntario se construyeron los perfiles plasmáticos y se calcularon los siguientes parámetros farmacocinéticos:
Análisis estadístico. Para el propósito de declarar bioequivalencia las principales variables analizadas fueron: ABC0ºº, Cmax, Tmax y ABC0Tmax. Los valores obtenidos de estas variables para los dos productos, se analizaron estadísticamente por medio del análisis de varianza (ANOVA), para determinar si se presentan diferencias significativas en los valores de las variables estudiadas, debidas a cada una de las fuentes de variación: productos, sujetos, períodos y secuencias de administración. Los valores de ABC0ºº, Cmax y ABC0Tmax se transformaron a logaritmo natural. El Tmax se analizó sin transformar16. Para determinar la bioequivalencia entre las formulaciones estudiadas se aplicó el método sugerido por Schuirmann17 y aceptado por la FDA, conocido como el “Método de dos pruebas unilaterales”, teniendo en cuenta el valor estándar del ANOVA calculado para cada parámetro. El nivel de significancia (a) para las pruebas o probabilidad de error fue 0.05 y se construyeron intervalos de confianza de 90% para el cociente entre las medias del producto de prueba y del producto de referencia. Criterio de aceptación. El producto de prueba se declaró bioequivalente con respecto al producto de referencia si, y sólo si los intervalos de confianza de 90% para el cociente de medias de los datos tanto del ABC0ºº como del Cmax transformados a logaritmos naturales están incluidos en el intervalo de 80% a 125% y el Tmax sin transformar está incluido en el intervalo de 80% a 120%. Los datos también fueron analizados en relación con los supuestos del modelo cruzado, dos períodos, dos secuencias, dos formulaciones para evaluar:
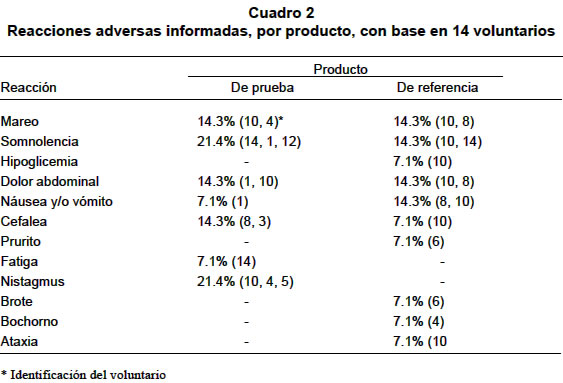
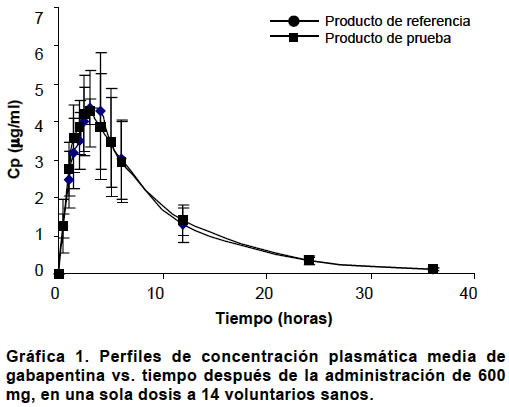
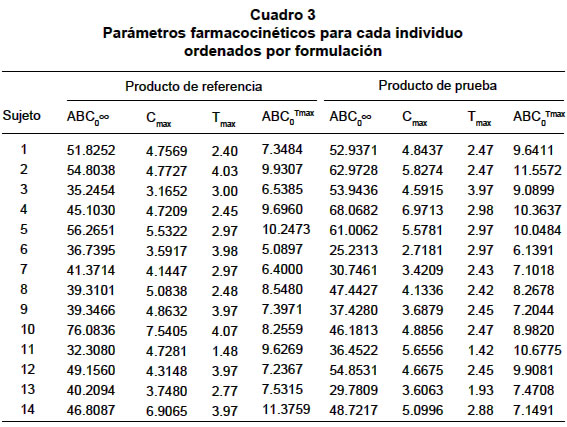
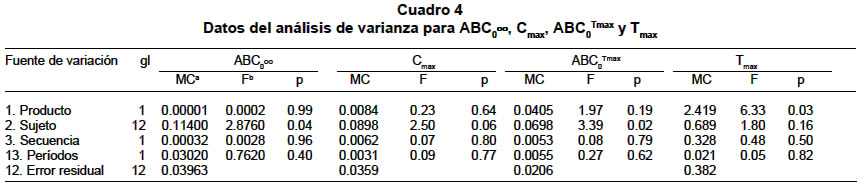
La diferencia se consideró estadísticamente significativa cuando el valor p<0.05. RESULTADOS Estudio de equivalencia farmacéutica. Las pruebas de contenido de gabapentina por cápsula y disolución permitieron concluir que los dos productos son equivalentes farmacéuticos (Cuadro 1). Estudio de bioequivalencia. Con ambos productos se presentaron reacciones adversas (Cuadro 2). No hubo deserciones ni retiros durante el estudio. Algunos voluntarios presentaron hasta tres reacciones adversas simultáneamente. Las concentraciones plasmáticas medias en mg/ml en cada tiempo de muestreo dieron origen a los perfiles plasmáticos medios para cada formulación los cuales se presentan en la Gráfica 1 y los parámetros farmacocinéticos para cada formulación y cada sujeto están consignados en el Cuadro 3. Los resultados del ANOVA, realizado a los parámetros farmacocinéticos ABC0ºº, Cmax y ABC0Tmax transformados a logaritmo natural y el Tmax original, se informan en el Cuadro 4. Los resultados obtenidos con respecto a los intervalos de confianza de 90%, expresados en porcentaje para el ABC0ºº, Cmax y ABC0Tmax para el producto de prueba, son en su orden: 87.5%-114.5%, 85.0%-109.7% y 97.9%-118.8%, los tres contenidos en el intervalo de confianza de 80% a 125% de la formulación de referencia. El intervalo de confianza para el Tmax del producto de prueba es de 68.4%-94.6% que no está contenido en el intervalo de confianza de 80% a 120% del producto de referencia. De acuerdo con las directrices de la FDA los dos productos son bioequivalentes con respecto ABC0ºº, Cmax y ABC0Tmax y no bioequivalente con respecto al Tmax. En la comprobación de los supuestos para el modelo ANOVA en el diseño cruzado, se obtuvieron los siguientes resultados:
DISCUSIÓN Los dos productos fueron equivalentes farmacéuticos, es decir que las pruebas fisicoquímicas y farmacotécnicas permitieron demostrar que los dos productos eran similares en cuanto a las especificaciones establecidas para esta forma farmacéutica, pero se comprobó que a pesar de ello, no se puede garantizar que su comportamiento en el organismo sea idéntico, porque el parámetro farmacocinético de mayor diferencia en este estudio fue el Tmax, para alcanzar la Cmax; la Tmax media del producto de referencia fue 3.18 horas y para el producto de prueba 2.59 horas. Esta diferencia significativa se puede explicar por la existencia de factores tales como el origen del principio activo, los excipientes utilizados, la tecnología empleada en la elaboración de la forma farmacéutica, que hacen que se demore en alcanzar la concentración plasmática máxima. Sin embargo, las variables ABC que permite medir la extensión de la absorción y la Cmax que representa el nivel de principio activo máximo que puede afectar la respuesta terapéutica del fármaco, se encuentran cercanos entre los dos productos, es decir que presentan una media y una desviación estándar que no difiere significativamente. Los resultados obtenidos al analizar el efecto de arrastre, período y formulación, permitieron considerar la bioequivalencia de las dos formulaciones aplicando los delineamientos sugeridos por la FDA con el ANOVA. La variabilidad intersujeto, interpretada como la respuesta de varios sujetos a la misma formulación, presentó en el ABC o sea en la extensión de la absorción, lo cual pudo ocurrir debido a factores fisiológicos del tracto gastrointestinal propios de algunos individuos. Estos hallazgos están en concordancia con los presentados por Gidal et al.4, en el que demostraron en dos estudios que la variabilidad intersujetos fue sustancialmente mayor que la intrasujetos para el ABC. Para que dos medicamentos sean declarados bioequivalentes, la FDA ha establecido que deben ser bioequivalentes en las variables farmacocinéticas ABC0ºº, Cmax y Tmax, sin considerar la duración del tratamiento en el que será utilizado, la frecuencia de administración y la respuesta esperada. En el caso de la gabapentina si se utiliza en un tratamiento de uso crónico la equivalencia con respecto al ABC y al Cmax garantizan que se mantendrá la extensión de la absorción y que se llegará a la concentración máxima que asegura la respuesta terapéutica esperada. CONCLUSIONES
AGRADECIMIENTOS Al profesor Abel Díaz sus aportes en el análisis estadístico del presente estudio. La financiación por Tecnoquímicas S.A., contrato 8716/01 de 2002, entre la Universidad de Antioquia y Tecnoquímicas S.A. Los autores aclaran que ninguno de ellos tiene ni ha tenido vínculos laborales o intereses personales con los laboratorios farmacéuticos Tecnoquímicas S.A. o Parke Davis que puedan derivar en conflicto de intereses. REFERENCIAS
Copyright 2004 - Revista Colombia Médica The following images related to this document are available:Photo images[rc04002g1.jpg] [rc04002t3.jpg] [rc04002t2.jpg] [rc04002t1.jpg] [rc04002t4.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}