 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Revista Colombia Médica, Vol. 35, No. 2, 2004, pp. 112-120 Proteínas celulares cómplices de las proteínas regulatorias y accesorias del VIH-1 María Eugenia Castaño, Bact.1, Silvio Urcuqui, Biol., M.Sc., Ph.D.2
Recibido para publicación septiembre 30,
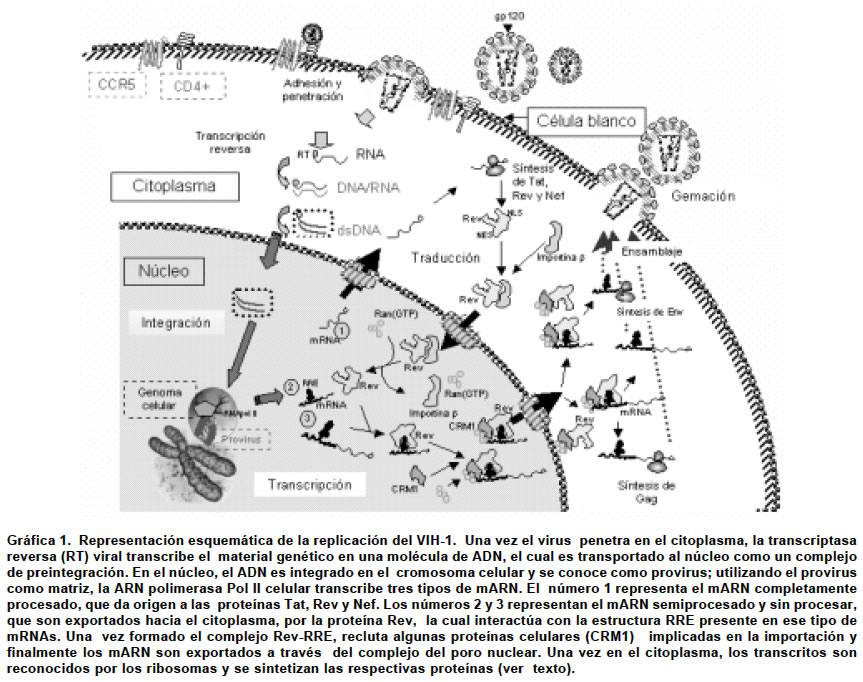
2003 Code Number: rc04018 RESUMEN La resistencia del virus de la inmunodeficiencia humana tipo 1 (VIH-1) a medicamentos antivirales ha presionado la búsqueda de métodos alternativos de terapia. En esta revisión, se presenta un análisis de los avances más recientes en el estudio de la interacción entre las proteínas regulatorias y accesorias del VIH-1 con factores celulares, sus efectos en la célula huésped y los beneficios para el virus. Se muestra cómo estas proteínas virales alteran procesos fundamentales de la célula huésped de manera muy precisa y las utilizan para aumentar la replicación viral, evadir la respuesta inmune del hospedero y causar enfermedad. Palabras clave: Virus de la inmunodeficiencia humana tipo 1. Proteínas. Patogénesis. SIDA. SUMMARY The resistance of human immunodeficiency virus type 1 (HIV-1) to antiviral drugs, has prompted a search for alternative therapy methods. This review presents an analysis of the most recent progress in the study of binding among the HIV-1 regulatory/accessory proteins and cell factors, their effects in the cell host and the benefits for the virus. Here, we discuses about how those viral proteins disturb fundamental host cell processes in a very precise way, how the virus uses its proteins to increase the viral replication, to evade the host immune response and to produce disease. Key words: Human immunodeficiency virus type 1. Proteins. Pathogenesis. AIDS. El virus de la inmunodeficiencia humana tipo 1 (VIH-1), el agente causal del síndrome de inmunodeficiencia humana (SIDA) es un retrovirus miembro del género de los lentivirus. Su genoma, además de presentar genes que codifican para las proteínas estructurales gag, pol y env, comunes a los otros retrovirus, codifica 6 proteínas accesorias/regulatorias Tat, Rev, Vif, Vpr, Vpu y Nef comprometidas en la regulación de la infección, producción viral y patogenicidad1-3. Durante la infección el VIH-1 utiliza múltiples factores celulares para su ciclo de vida y establece una interrelación con la célula y el hospedero; incluso se vale de algunas proteínas celulares para causar enfermedad (patogénesis) y como herramienta para favorecer la infección. En la infección por VIH-1, tanto in vivo como in vitro, se afecta un gran número de funciones de las células del sistema inmune, incluyendo la quimiotaxis, fagocitosis, muerte intracelular y producción de citoquinas en los monocitos/macrófagos. Un claro ejemplo es la modulación por parte de las proteínas virales Nef, Vif, Vpu, Vpr y Rev de VIH-1, de un número importante de vías de señalización a través de interacciones con el citoesqueleto y proteínas citoplásmicas4. Otro ejemplo de cómo las proteínas virales afectan la célula hospedera es la regulación de la expresión en la membrana celular de receptores como el complejo mayor de histocompatibilidad tipo I (MHC-I), regulado por tres proteínas virales en diferentes niveles, la proteína Tat que inhibe la transcripción de MHC-I, Vpu que retiene las cadenas peptídicas nacientes de MHC-I en el retículo endoplásmico y Nef que media la internalización selectiva de moléculas desde la membrana plasmática5. Tat, un inductor de la transcripción de genes celulares. El alto nivel de transcripción del ADN del VIH-1 integrado en el genoma del hospedero (provirus), es regulado por una proteína viral de aproximadamente 14 kD llamada Tat. Tat es un activador secuencia específico inusual que reconoce un ARN altamente estructurado presente en los transcritos nacientes, lo que permite estimular la transcripción del provirus. En ausencia de Tat en las células infectas las proteínas celulares inician la transcripción del genoma del VIH-1, pero la mayoría de transcritos sólo alcanzan entre 50 y 60 nucleótidos, que luego son degradados. Esto significa que la transcripción de todo el genoma viral es muy deficiente. Una vez se ha sintetizado la proteína Tat a partir de un primer ARNm viral completamente procesado, ésta interactúa con la estructura TAR ARN presenten en los transcriptos nacientes, activa la elongación de los transcritos virales y por ende, una eficiente transcripción de la totalidad del genoma viral. Por tanto, la proteína Tat es un factor transcripcional, con un dominio de activación en la región central que funciona en forma independiente del dominio de unión a ARN4,6. La actividad de la Tat es dependiente de la estructura TAR (por Tat Activation Region), presente en el extremo 5’ de los ARN virales nacientes. Si bien la transcripción del VIH-1 es mediada por la ARN polimerasa II celular, la función principal de Tat es a nivel de la elongación de la transcripción. Por tanto, la Tat juega un papel fundamental en la regulación de la transcripción total del genoma y de la replicación del VIH-14. En este artículo se tratará en primer lugar el papel de Tat en la transcripción del genoma viral y luego su función en la patogénesis. Tat en la transcripción viral. Varios investigadores identificaron un complejo proteico celular llamado TAK (Tat Associated Kinase) que se une al dominio de activación de Tat una vez que ésta interactúa con su ligando Tar y permite la formación de un complejo capaz de fosforilar el dominio carboxiterminal (CTD) de la subunidad mayor de la ARN polimerasa II (ARNPII)4,7. Se demostró que TAK era el mismo complejo que previamente se había llamado PITALRE, expresado de manera ubicua en tejidos humanos, implicado en la elongación de la transcripción. PITALRE había sido renombrado Cdk9 porque se relacionaba con la familia de quinasas dependientes de ciclinas (Cdks). Por el papel que desempeña Cdk9, se buscó una ciclina que pudiera conferirle a TAK especificidad por su sustrato y se identificó la ciclina T (CykT). Luego se determinó que CykT aumenta la especificidad de Tat por TAR, lo que incrementa su actividad y es necesario para la elongación de los transcriptos in vitro4. Este complejo también se conoce como factor positivo de elongación de la transcripción b (P-TEFb)6,7. Por análisis de secuencia de aminoácidos, se especuló que PITALRE y P-TEFb eran homólogas o que P-TEFb formaba parte de PITALRE6. Aunque se han descrito otros factores que interactúan con Tat-TAR y que posiblemente contribuyen a la transcripción de VIH-1, la interacción de Tat y TAR con Cdk9-CycT parece ser crítica en la progresión de la transcripción8. También se ha descrito la interacción de Tat con otros factores de transcripción incluyendo los factores de transcripción IIH (TFIIH) e IIF (TFIIF). Gracias a estas interacciones, Tat estimula la fosforilación del dominio CTD de la RNAPII. La fosfatasa FCPI es capaz de defosforilar el dominio CTD de la RNAPII; sin embargo, algunos investigadores7 han descrito que gracias a la interacción Tat-FCPI se inhibe dicha defosforilación. Tat también recluta a la enzima responsable del encapuchamiento del mARN de mamíferos (Mce1) y a la metiltransferasa cap (Hcm1). El reclutamiento de estas enzimas hacia TAR aumenta la eficiencia de formación del encapuchamiento en el ARN transcrito9,10. Ambas, en asocio con la ARNPII dependiente de la fosforilación de CTD, trabajan en la elongación de los transcriptos. La modificación postranscripcional de encapuchamiento del mARN de VIH-1, es fuertemente estimulada por la región C-terminal de Tat, que interactúa directamente con Mce1. También se ha descrito que Tat puede ser acetilada al interactuar con diferentes tipos de histona acetil transferasa (HATs), incluyendo el complejo CBP/p300, PCAF y Hgcn58,11, que afecta la expresión génica tanto viral como celular12. En resumen, gracias a la capacidad del complejo Tat-TAR de interactuar con quinasas celulares, promueve la transcripción de mARN virales completos, que luego son transportados al citoplasma para la expresión de las proteínas virales necesarias para producir una nueva progenie viral infecciosa7. Tat y patogénesis. Se ha demostrado que Tat se une con Egr-2 y 3 y aumenta la transactivación del promotor de Fas ligando (FasL) mediada por Egr; esta sobre-regulación de FasL podría contribuir a la apoptosis de células T durante la infección por VIH-17. Por el contrario, Zhang M. et al.13 lograron determinar que la infección por VIH-1 sobre-regula tanto los niveles de mARN de Bcl-2 como de la proteína misma, empleando un sistema de cultivo de macrófagos derivados de monocitos humanos; al regular la producción de esta proteína antiapoptótica podría estar protegiendo las células infectadas de la muerte por esta vía. La interacción de Tat con la Mn-superóxido dismutasa (Mn-SOD) también parece tener un efecto en la patogénesis de VIH-1. Tat reprime la expresión de Mn-SOD al elevar el estrés oxidativo, lo que aumenta la proliferación celular, la apoptosis o altera la actividad de proteínas que contienen tiolato de zinc, como Sp17. Tat interactúa además con Pur a, una proteína celular comprometida en la transcripción. Esta interacción está ligada con la activación de virus que causan infecciones oportunistas en pacientes con SIDA y con la transactivación de LTR de VIH-1. La interacción de Tat con el factor de transcripción NFAT1, PKC y con el complejo cooperativo rel/AP1 puede estar implicada en la alteración de la producción de citoquinas como IL-10 e IL-27. La exposición de cultivos celulares de diferente origen (sistema nervioso central y células T) con extractos altamente purificados de Tat, eleva la actividad transcripcional de constructos reporteros que contienen promotores del factor de crecimiento transformante b 1 (TGFb-1), del factor de necrosis tumoral a (TNFa) y el LTR del VIH-1; además de esto, el tratamiento resultó en un aumento en el nivel de mARN de TGFb-1 y TNFa en estas células14. Otros investigadores confirman estos hallazgos al informar que durante la progresión a SIDA, se eleva la expresión de varias citoquinas comprometidas en la transducción de señales tales como TGFb-1, y de citoquinas proinflamatorias incluyendo MCP-115. El aumento de estas citoquinas es mediado, al menos en parte, por la proteína Tat del VIH-1. La inhibición de la expresión de MHC-I mediada por Tat probablemente se debe a una interacción directa con TAFII250, una subunidad de TFIID que impide que éste se una a la caja Sp1, interacción indispensable para la actividad del promotor de MHC-I5. Se ha descrito también que Tat es capaz de interactuar con otras proteínas celulares, pero se desconoce si esta interacción está relacionada o no con alguna función específica entre otras la interacción con Tip110, con la proteína de unión a Tat 1, con el factor estimulador de Tat 1 (Tat-SF1), con el dominio POZ de la proteína FBI-1, con el receptor nuclear coactivador de la proteína GRIP1, proteína GLI-2, con el factor de elongación Spt5 y con la proteína de unión a ADN/ARN de cadena sencilla YB-17. Vpr: una máquina de control del ciclo celular. El VIH-1 tiene la particularidad de replicarse en células que no están en división activa. La proteína viral Vpr, de aproximadamente 14 kDa, es probable que juege un papel muy importante en la inhibición de la expansión clonal al permitir la detención de la célula en la fase G2 del ciclo celular, etapa donde el LTR del VIH-1 es más activo4,16. Sólo la expresión de Vpr es suficiente para alterar el ciclo celular. Además, Vpr es un transactivador débil del LTR del VIH-1 y de otros promotores virales homólogos, como el del virus Epstein Barr y el de citomegalovirus17. Sherman et al. 19, caracterizaron a la Vpr como una proteína nucleocitoplásmica con dos señales de importación y una señal de exportación nuclear dependiente de exportina-1 y nucleoporina. Mutaciones en el NES de la Vpr debilitan su incorporación en el nuevo virión4,18. También ellos demuestran que la secuencia de exportación nuclear (NES) de Vpr es requerida para una eficiente replicación de VIH-1 en macrófagos. Se ha demostrado que virus naturalmente no infecciosos o virus defectuosos gracias a inhibidores de la transcriptasa reversa o proteasa, son capaces de parar el ciclo celular; esos resultados sugieren que tanto los virus infecciosos como los no infecciosos in vivo así como aquellos circundantes de las células dendríticas foliculares participan en la supresión inmune, gracias a la acción de Vpr20. Vpr induce la diferenciación celular incluyendo la activación de la transcripción de genes celulares; induce la inhibición del crecimiento de cultivos celulares primarios y varias líneas celulares de origen tumoral, aún en ausencia de otras proteínas virales, mediante un bloqueo del ciclo celular en la fase G2/M21. En 1998 se identificó un factor celular que interactúa con Vpr, denominado hVIP/MOV34 (proteína humana que interactúa con Vpr) que es idéntico al mov34, una proteína perteneciente a la familia de factores reguladores de la transcripción, miembros de la familia de proteasoma y proteínas celulares comprometidas con la transición de la fase G2/M del ciclo celular en mamíferos22. En este mismo trabajo demuestran una asociación entre la inducción de parada del ciclo celular en G2/M inducida por Vpr y un cambio en la localización subcelular de hVIP/MOV34 desde el núcleo a una localización perinuclear. El proceso está asociado con una inhibición de la maduración promovida por factores asociados con la actividad quinasa de la histona H1. Ramanathan et al.23 demuestran que la región carboxiterminal de hVIP es crítica para su interacción con Vpr. Mediante ensayos de inmunoprecipitación, Sawaya et al.24 lograron determinar que la Vpr interactúa con Sp-1 y con P53, posiblemente formando un complejo tripartita; P53 es otro elemento clave en el control del ciclo celular. Vpr y la importación hacia el núcleo. El complejo de preintegración de VIH-1 (VIH-1 CPI) entra en el núcleo atravesando el canal acuoso central del complejo del poro nuclear. El VIH-1 CPI presenta tres proteínas nucleofílicas, matriz, integrasa y Vpr. Parece ser que la Vpr actúa en una faceta complementaria para aumentar la importación del CPI al núcleo celular en ausencia de división25. Ensayos de colocalización celular realizados por Vodicka et al.16 mostraron que Vpr se encuentra en el complejo de poro nuclear (NPC), con Nsp1p y con Nup159p. Describen que Vpr no sólo colocaliza con la importina-a y Nsp1p, sino que interactúa con ellas; sugieren además que la localización nuclear de Vpr obedece a su tamaño pequeño más que a su capacidad de entrar al núcleo, pues la proteína heteróloga ß-galactosidasa fusionada con Vpr no pudo ser tanslocada al núcleo por esta proteína viral. Vpr puede interactuar específicamente con la región FG (repeticiones de fenilalanina-glicina) de la nucleoporina Pom12118, que forma parte del CPN. Interacción de Vpr con otras proteínas celulares. Se ha descrito que Vpr interactúa con la uracil ADN glicosilasa (UNG), una enzima de reparación comprometida con la remoción del uracilo en el ADN, pero no afecta su actividad enzimática1. Estudios in vitro han mostrado que la Vpr se une con la proteína citosólica Rip-1 e induce su translocación al núcleo17; posiblemente Rip-1 es una proteína transportadora que lleva a Vpr al núcleo. Además, Vpr y Rip-1 coinmunoprecipitan con el receptor de glucocorticoides humano como parte de un complejo activado de receptor. Estos datos podrían incluir a Vpr en la vía transcripcional mediada por el receptor de glucocorticoide. Rev, proteína viral crucial en la exportación de transcriptos de VIH-1 sin procesar. La proteína viral Rev es funcional en una amplia variedad de células eucariotes, incluyendo levaduras, Drosofila, huevos de Xenopus y mamíferos. Esto sugiere que los cofactores celulares que interactúan con Rev podrían ser proteínas conservadas evolutivamente, esenciales para la función de células normales3. Similar a Tat, la proteína Rev participa en la regulación de la expresión del genoma de VIH-1, es decir, se requiere de Rev para la expresión de la mayoría de las proteínas de VIH-1 necesarias para el ensamble de partículas virales infecciosas3. VIH-1 una vez integrado al genoma (estado proviral) genera tres tipos diferentes de mARN según su procesamiento: de 9 kb (no procesados) que codifica para las proteínas gag y gag/pol; de 4 kb (semiprocesados) que codifican para env, vif y vpu, y de 2 kb (completamente procesados), que codifican para las proteínas Tat, Rev y Nef26; en consecuencia, todos estos transcriptos son necesarios para una eficiente síntesis de proteínas virales en el citoplasma. Es de anotar que la mayoría de los ARNs celulares sin procesar son retenidos en el núcleo, donde se maduran o degradan; por tanto, los mARNs virales de 9 y 4 kb, deben ser exportados al citoplasma por mecanismos adicionales3,27. Para resolver este problema, el genoma del VIH-1, cuenta con la proteína Rev, que tiene la capacidad de interactuar con la estructura RRE (Rev Response Element) presente en los transcriptos virales que muestran intrones y los exporta al citoplasma en asocio con ciertas proteínas celulares, a través del CPN28. Rev específicamente induce la acumulación de estos transcriptos en el citoplasma27. La proteína Rev, de aproximadamente 18 kDa, consiste de un dominio aminoterminal rico en arginina, que participa en tres funciones diferentes: interactuar con RRE, en la homodimerización que se produce en respuesta la unión con el RRE y como una señal de localización nuclear (NLS). En la región carboxiterminal presenta un dominio efector rico en leucina, que funciona como una NES, dependiente de la exportina 1. La nucleoporina humana hRIP/Rab interactúa específicamente con el dominio efector de Rev27. Los resultados obtenidos por Fritz et al. sugieren que el dominio efector de Rev mimetiza las NES de las proteínas celulares porque la NES de PKI puede reemplazar funcionalmente el dominio efector de Rev, gracias a su interacción con hRIP/Rab27. El papel de PKI en la célula todavía no es claro, pero esta proteína puede entrar al núcleo e inhibir las funciones nucleares de la proteína quinasa dependiente de cAMP. Venkatesh et al.29, demostraron que la REBP (Rev/Rex effector binding protein) es capaz de aumentar la expresión de un gen reportero dependiente del complejo Rev-RRE, ya sea en forma independiente o en forma cooperativa con hRIP/Rab. Además, los autores sugieren que REBP podría funcionar como un mediador de la función de NES de Rev y participar en la exportación del ARN durante la infección viral. Translocación citoplasma-núcleo. La proteína Rev es translocada hacia el núcleo gracias a la NLS presente en la región C-terminal de la proteína, que es reconocida por la importina a (también conocida como carioferina a), que a su vez se une a un segundo factor de importación, la importina ß (o carioferina ß) (Gráfica 1). Se ha demostrado que en el ingreso de Rev al núcleo también participa la fosfoproteína B23 que interactúa con la NLS28,30. También se ha descrito que moléculas de ARN son capaces de inhibir la importación de Rev hacia el núcleo en células permeabilizadas31, gracias a la inhibición de la interacción entre el dominio rico en arginina de Rev y la importina ß. Exportación de transcriptos virales semimaduros y sin madurar. El mecanismo molecular para la exportación nuclear mediada por Rev ha sido objeto de intenso estudio, lo que ha permitido comprender muchos procesos utilizados por la misma célula y profundizar en el conocimiento de la relación virus-hospedero. Una vez se identificó a Rev como la proteína responsable de la exportación de los transcriptos virales, muchos grupos se dieron a la tarea de encontrar las proteínas celulares implicadas en esta función. La primera proteína identificada fue la exportina 1. El análisis de la secuencia primaria de la exportina 1/Crm1 demostró que es miembro de la superfamilia de importina-b (karioferina-b), receptores comprometidos en el transporte nuclear4. En el núcleo, una vez Rev se une a mARN que contiene RRE (Gráfica 1), al menos dos proteínas solubles celulares se asocian con este complejo: CRM1 y Ran, para formar el complejo de exportación. También se ha demostrado que la NES de Rev interactúa cooperativamente in vitro con la exportina 1 y Ran guanosina trifosfato (GTPasa), un factor esencial de transporte nuclear, de una manera que requiere que Ran esté en su forma unida a GTP4. La primera asociación entre Rev y la exportina1/Crm1 se realizó usando la citotoxina leptomicina B (LMB), porque inhibe la exportación de mARN dependiente de una NES en cultivos celulares, sugiriendo que el complejo Rev/RRE-CRM1-RanGTP es sensible a esta citotoxina32. Este complejo terciario (Rev-RRE-CRM1) interactúa con algunas nucleoporinas presentes en el complejo del poro nuclear, lo que al final resulta en la translocación del mARN viral sin procesar del citoplasma. Mediante el sistema de doble híbrido en levadura, se identificaron 2 grupos de proteínas que interactúan con Rev en forma dependiente de la NES: hRip (human Rev-interacting protein) y Rab (Rev/Rex activation domain-binding protein)28; hRIP es una proteína que forma parte de la vía de exportación de proteínas del núcleo al citoplasma. La sobre-expresión de hRIP/Rab en células de mamífero, aumenta la actividad de Rev27. Se ha demostrado que diferentes proteínas se unen al dominio de activación de Rev, entre ellas Rip1p (proteína celular de levadura que hace parte del complejo del poro nuclear) y su homólogo en mamíferos, hRIP/Rab y el factor de iniciación eucariótico 5A (eIF-5A), cuya interacción interviene en la exportación de los transcriptos virales sin madurar27,33,64. Rev y el procesamiento de mARN. Se han descrito otros factores celulares que interactúan con Rev. Las proteínas YL2 (murina) y su homólogo humano p32 interactúan con el dominio básico de Rev (el mismo dominio que interactúa con RRE) y contribuye al proceso de oligomerización3. La proteína p32 se asocia con ASF/SF2, un factor esencial en el splicing y se ha hipotetizado que contribuye como un puente con la maquinaria celular de splicing3. Powell et al.35 demostraron que ASF/SF2 (miembro de la familia de proteínas serina-arginina -SR- comprometida en la regulación del procesamiento del mARN) interactúa específicamente con una subregión de RRE in vitro, de manera dependiente de Rev; la sobreexpresión de SF2/ASF y SC35 inhiben la función de Rev y la replicación viral36. Esos resultados sugieren que en la inhibición del procesamiento de los transcriptos virales Rev induce un salto de los exones gracias a una interacción simultánea de Rev y proteínas SR con RRE, lo que altera el subsecuente ensamblaje y actividad catalítica del complejo de procesamiento. También se ha demostrado que la interacción de Rev con hTra2a (miembro de la familia SR) estimula las funciones de Rev, sin alterar o bien aumentando la concentración de ARN26. Vpu y Nef modulan la señalización celular. La glicoproteína de envoltura ENV se une al receptor celular CD4 no sólo en la superficie celular sino también en el retículo endoplásmico (RE) antes de la translocación de la proteína viral con la membrana plasmática37. Por esto, el genoma de VIH-1 codifica tres proteínas, Vpu, Nef y gp120, que regulan negativamente la expresión de CD4 en la superficie de la célula durante la infección viral para expresar así sus glicoproteínas de envoltura en la membrana celular. VIH-1 ha desarrollado estrategias tanto para remover su receptor CD4 de la superficie (Nef) como para evitar que el receptor recién sintetizado llegue a la superficie celular (Vpu). Se ha informado además que tanto Vpu como Nef regulan negativamente la expresión del complejo mayor de histocompatibilidad tipo I38. Así, VIH-1 ha desarrollado diversos mecanismos para perturbar el tráfico intracelular de proteínas del hospedero con el propósito de aumentar la virulencia de la partícula viral y escapar de la vigilancia inmune. Vpu como regulador. Vpu es una proteína de aproximadamente 16 kDa, con un dominio hidrofóbico de anclaje a la membrana y una cola citoplásmica polar fosforilada. Se han establecido dos funciones principales para Vpu: degradación de CD4 y aumento en la liberación de las partículas virales de la membrana plasmática38. Vpu se une con CD4 en el RE y lo hace blanco de proteolisis en el citosol por la vía ubiquitina-proteasoma39,40. La conexión directa entre Vpu y el proteasoma se estableció demostrando que Vpu se une a la proteína celular b-TrCP, que a su vez se une con el factor blanco del proteasoma Skp1p. b-TrCP interactúa con Vpu y su reclutamiento en la membrana requiere de la fosforilación de los residuos serina 52 y serina 56 presentes en el motivo de fosforilación DSGXXS de Vpu, esenciales para la degradación de CD441; la responsable de esta fosforilación es la caseína quinasa II (CKII)40,42. La interacción de Vpu con b-TrCP se realiza a través de regiones WD repetidas que se encuentran en la región C-terminal de b-TrCP y gracias a una caja F presente cerca de la región N-terminal implicada en la interacción con SKPIP. Mutaciones de la caja F de b-TrCP tienen un efecto dominante negativo sobre la degradación de CD4 mediado por Vpu40. b-TrCP es un componente del complejo ligasa ubiquitina E3. La importancia de estas interacciones se confirmó con el aislamiento de un complejo terciario CD4-Vpu-b-TrCP in vivo39. Esta interacción regula la degradación de IkB-a, teniendo efecto al final en los genes regulados por NF-kB y en la inducción de apoptosis43. También se ha descrito que Vpu inhibe la actividad de NF-kB interfiriendo con la degradación mediada por b-TrCP de IkB-a42. Además de esta función, se ha descrito que Vpu puede disminuir la expresión de MHC-I en la membrana citoplasmática5,38,44. Esto significa que al degradar los CD4 y afectar el MHC-I, Vpu permite por un lado preservar la infectividad viral y al mismo tiempo promueve la evasión del sistema inmune. Nef, otro factor de virulencia. Entre los productos génicos de VIH-1 implicados en la modulación de la señalización celular, Nef parece ser el más potente. Se considera el factor principal de virulencia tanto in vivo como in vitro45. Nef es una pequeña proteína de 27 kDa, requerida tanto para la replicación máxima del virus como para la progresión de la enfermedad, mediante el control de los factores de señalización de la célula46. Se ha demostrado que defectos y variaciones en el gen que codifica para esta proteína, son responsables de una rápida, lenta o no progresión del SIDA47. Nef es una proteína de 206 aminoácidos con grupos miristilo, que se expresa de forma abundante durante la infección por VIH-1 y se localiza en la membrana plasmática, el citoplasma, el núcleo y dentro del virión46. Esta miristoilación en la región aminoterminal de Nef y el dominio de unión a SH3 rico en prolina, sugieren que esta proteína podría interactuar con proteínas del hospedero en la membrana plasmática45. Nef además remueve CD4 que ya está en la superficie celular, al acelerar la endocitosis a través de vacuolas cubiertas de clatrina48. Algunos autores sugieren que la endocitosis ocurre a través de interacciones entre Nef y un complejo proteico, el complejo adaptador AP-2, que recluta proteínas transmembrana a vacuolas recubiertas con clatrina. Específicamente, se encontró que Nef colocaliza con el complejo AP-2 en la membrana plasmática y que se une a sus subunidades directamente48. Nef se une al complejo de proteínas adaptadoras (AP) de las vesículas cubiertas, induciendo la expansión del compartimento endosomal y alterando la expresión en la membrana celular de proteínas como CD4, CD28 y MHC-I; es decir, Nef al igual que Vpu, es capaz de regular negativamente la expresión de estos receptores para alterar el estado de activación de las células T, aumentando así la infectividad de la partícula viral49. Aunque son algo confusos los aspectos específicos de la alteración de la cascada de señalización de células T por Nef, se ha descrito que Nef altera la vía de señalización de los receptores de células T (TcR), lo cual es crítico en la patogenicidad del SIDA50, la señalización a través del receptor de IL-2 y las vías que llevan a la producción de quimioquinas-monoquinas en macrófagos, así como múltiples cascadas anti-apoptóticas46. Concomitante con la alteración de la vía de activación de TcR, se presenta una sobre-regulación del factor de muerte Fas ligando, lo que le permite a la célula infectada entrar en contacto con células no infectadas e inducir su muerte46. Algunos estudios sugieren que Nef afecta la vía de activación de Fas a través de la inactivación de las caspasas 3 y 846. El efecto de Nef sobre el MHC-I tiene como propósito proteger las células infectadas del reconocimiento de los linfocitos T citotóxicos51, mediante una modulación negativa de HLA-A y B, cuya permanencia evita el reconocimiento por parte de las NK. Williams et al.51 muestran por primera vez que Nef funciona como un adaptador capaz de unir el MHC-I con proteínas implicadas en el tráfico celular. El mecanismo comprometido en la regulación de MHC-I por Nef sugiere que esta proteína viral, en asocio con PACS, arrebata la vía endocítica de ARFG, por un proceso dependiente de PI3K y regulación negativa del MHC-I, de la superficie celular del sistema trans-golgi52. En este proceso están involucrados tres motivos de Nef: una región ácida, un dominio SH3 y un motivo M, que controlan la salida de PACS-1 del sistema de trans-golgi, la activación de ARF6 y el secuestro en la parte interior del sistema trans-golgi de MHC-I respectivamente. Se ha descrito además que Nef estabiliza la unión de AP-1 y AP-3 con las membranas46,53 y que se co-localiza con la clatrina y con AP-1 a nivel del sistema trans-golgi, al igual que MHC-I, lo que sugiere que a este nivel también regula su expresión46. Nef en asocio con las quinasas celulares. Se ha demostrado que Nef in vitro se asocia por lo menos con una quinasa serina/treonina celular2. Nef interactúa además con la quinasa Src Lck de linfocitos T, produciendo daño específico en la señalización54. Nef es capaz de modificar la actividad catalítica de otros miembros de la familia Src, incluyendo Fyn, Hck y Lyn al interactuar con ellas46,54. Se ha descrito la interacción de Nef con otras proteínas celulares. La familia de proteínas PAK juega un papel muy importante en la organización del citoesqueleto y la apoptosis46. Nef interactúa con PAK-2, y se cree que participa en el ensamble y liberación de los viriones del VIH-1. Wolf et al.55 muestra que Nef es capaz de activar PAK, que compromete la fosforilación de la quinasa fosfatidil inositol-3 (PI-3). La asociación de Nef/PI-3/PAK fosforila e inactiva la proteína pro-apoptótica Bad, sin involucrar la proteína quinasa B-Akt-quinasa, lo que le permite inducir la muerte de las células vecinas sanas, pero protege las células infectadas de apoptosis por vía mitocondrial46. Nef también interactúa con P53, que termina en una disminución de la vida media de esta proteína proapoptótica, disminuyendo su unión con el ADN46. También se ha descrito la unión de Nef con miembros de la familia de quinasas serinatreonina, tales como MAPK y PKC. En efecto, se demostró que Nef es sustrato de PKC46 y que inactiva a MAPK56, pero aún es desconocido el papel o implicaciones de estas interacciones. Vif supervisa el ensamble del virus. La proteína viral Vif, al igual que las proteínas celulares ciclofilina A (CyPA) y la proteína-quinasa activada por mitógenos (MAPK) debe estar presente en la célula infectada donde se está llevando a cabo el ensamblaje de los viriones. La ausencia de estas proteínas resulta en el bloqueo de la infección poco después de la entrada en la célula blanco4. Vif es una proteína básica de 23 kDa que se produce en un estado tardío del ciclo de replicación; se ha demostrado que los residuos básicos de su porción carboxiterminal se asocian con la fase citoplásmica de la membrana celular57. Una fracción sustancial de Vif está presente en el sitio de ensamble del virus cerca de la membrana plasmática y se ha sugerido que esto capacita a Vif para modular el ensamble del virión de una manera que facilita el desensamble u otros eventos tempranos de infección4. Estudios recientes sugieren una asociación de Vif con la proteína intermedia de filamento, con la vimentina, con la tirosina quinasa Hck, y con HP68, una proteína celular de unión a ATP58; el papel de estas interacciones aún es materia de estudio. Se demostró por microscopía confocal que Vif se encuentra soluble en el citoplasma o se asocia con el citoesqueleto con los filamentos intermedios, vimentina y queratina57. La presencia de Vif afecta la estructura de los filamentos intermedios y puede producir colapso completo de la red conformada por ellos, resultando en la formación de agregados perinucleares que contienen tanto Vif como filamentos intermedios. CEM15/APOBEC36 es una proteína celular requerida para la resistencia a la infección por un VIH-1 deficiente del factor de infectividad Vif. Harris et al.59 muestran que esta proteína es una deaminasa incorporada en el virión durante la producción viral y tiene como función la deaminación de deoxicitidina a deoxiuridina en la cadena negativa de un cADN de retrovirus, produciendo la destrucción del virus. Los autores demuestran que Vif es capaz de proteger al MLV de ese efecto. CONCLUSIONES Para cumplir sus funciones las proteínas reguladoras y las accesorias necesitan interactuar con factores de la célula huésped en los diferentes pasos de la infección viral. Una coordinación mediada por la interacción proteína-proteína es clave para una replicación eficiente y a la vez para controlar la maquinaria celular. Sin embargo, a pesar de los avances logrados en los últimos años en la comprensión de la relación virus-hospedero, es necesario profundizar en el tema con el propósito de utilizar estos conocimientos como una herramienta en la lucha contra la dispersión del virus y/o el desarrollo o progresión de la enfermedad causada por la infección. En los últimos años, las proteínas virales regulatorias y/o sus ligandos, han comenzado a ser blanco de muchos estudios con fines terapéuticos, porque el ciclo de vida del virus es dependiente de su función. El desarrollo de péptidos o la caracterización de proteínas celulares capaces de inhibir en forma específica sus funciones, será un gran avance en la búsqueda de nuevos medicamentos que permitan luchar contra la progresión del SIDA. Igualmente, las características de las proteínas virales las convierten en un blanco potencial para estudiar los eventos celulares; un buen ejemplo es la proteína Vpr, que por su capacidad de retardar la entrada en fase G2, puede permitir estudiar eventos relacionados con el ciclo celular. También es muy interesante el mecanismo utilizado por Nef para regular negativamente la expresión de MHC-I en la membrana citoplasmática de la célula infectada. Mediante esta modulación, el VIH-1 logra escapar de la actividad de los linfocitos T citotóxicos (CTL). Se ha encontrado que el HLA-A, sobre todo el HLA-A2 y el HLA-B son regulados negativamente, mientras que HLA-C y HLA-E no se afectan; esta estrategia le permite a la vez al VIH-1 proteger a la célula infectada de la lisis por células NK. Este es un claro ejemplo de cómo el VIH-1 logra un equilibrio con el sistema inmune del hospedero para evitar ser eliminado y lograr así mantener la infección. En resumen, el genoma del VIH-1 presenta toda la información necesaria para obtener un arsenal que pone en “jaque mate” al hospedero, de manera regulada y sin medir las consecuencias REFERENCIAS
Copyright 2004 - Revista Colombia Médica The following images related to this document are available:Photo images[rc04018g1.jpg] |
| |||||||||
{kind=link}