 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Revista Colombia Médica, Vol. 35, Num. 4, 2004, pp. 191-198 Deleciones en el gen de la distrofina en
62 familias colombianas: correlación genotipo-fenotipo para la
distrofia muscular de Duchenne y Becker
Claudia T. Silva, Biol., M.Sc.1, Dora Fonseca, Biol., M.Sc.2, Carlos Martín Restrepo, M.D., M.Sc.3, Nora C. Contreras, Biol.2, Heidi E. Mateus, M.D.2 1. Instructora Asociada, Profesora Distinguida,
Unidad de Genética, Instituto de Ciencias Básicas, Facultad
de Medicina, Universidad del Rosario, Bogotá. e-mail: ctsilva@urosario.edu.co Recibido para publicación mayo 2, 2004 Code Number: rc04040 RESUMEN Introducción:La correlación genotipo-fenotipo se estableció mediante
el análisis de deleciones del gen de la distrofina en pacientes con
distrofia muscular de Duchenne y Becker (DMD/DMB). Palabras clave: Distrofia muscular Duchenne y Becker (DMD, DMB); Reacción en cadena de la polimerasa múltiplex (PCR); Corrimiento del marco de lectura; Herencia ligada con la X recesiva. SUMMARY Introduction: A genotype-phenotype correlation was established by dystrophin gene
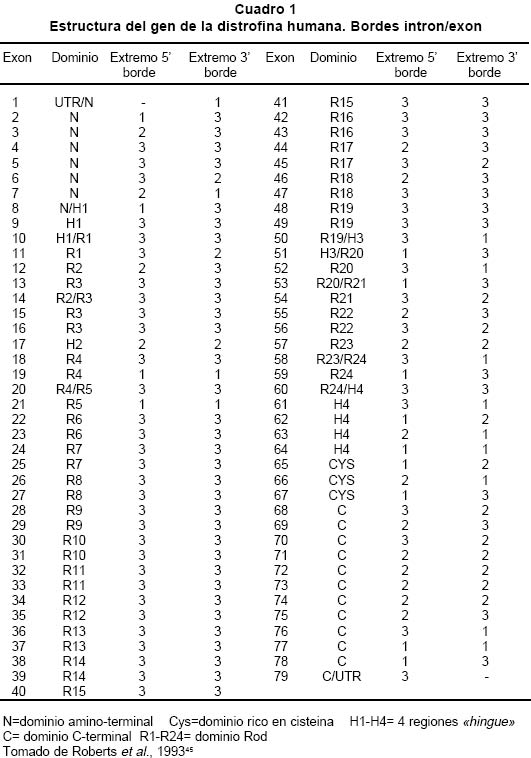
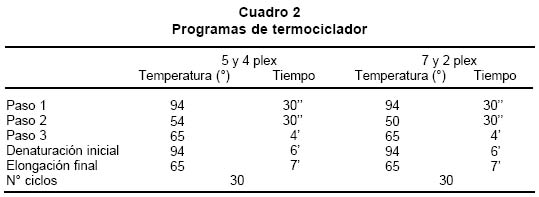
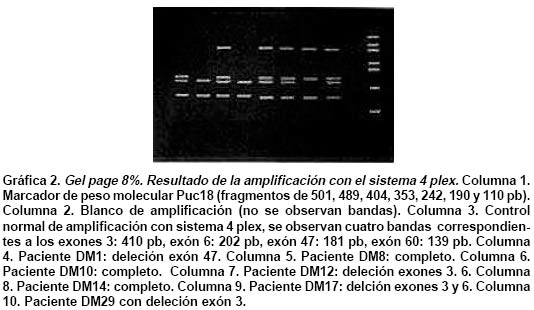
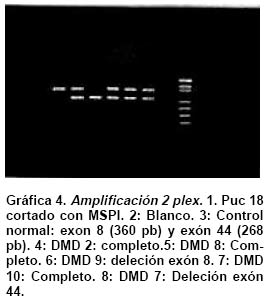
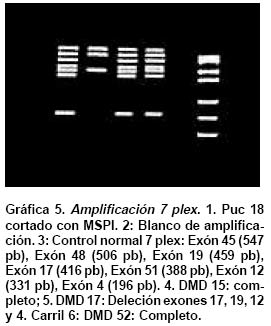
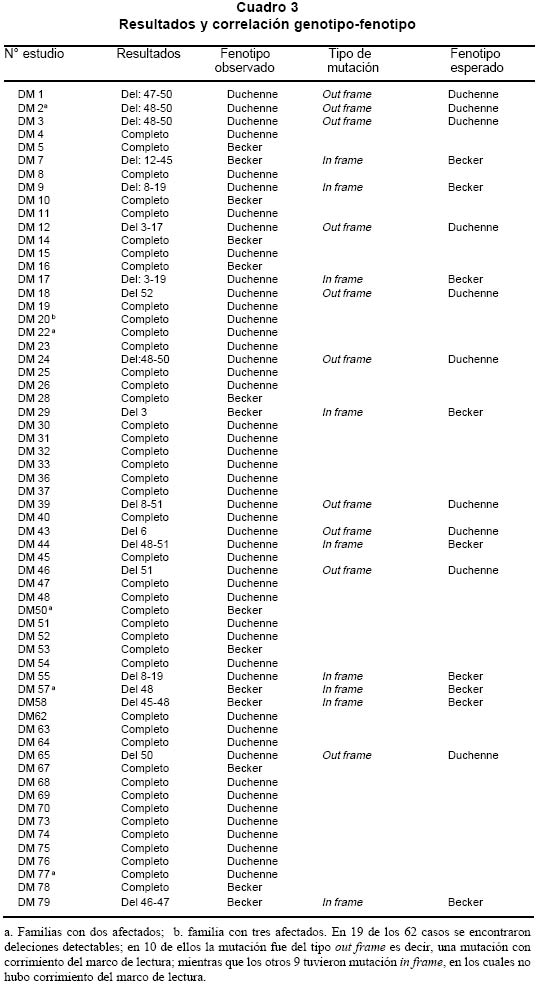
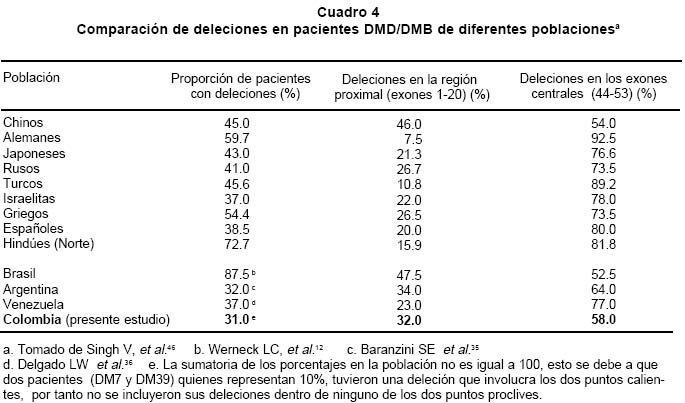
deletion analysis in Duchenne and Becker muscular dystrophies patients (DMD/BMD). Key words: Duchenne and Becker muscular dystrophy (DMD/DMB); Multiplex polymerase chain reaction (PCR); Open reading frame hypothesis; X-linked diseases. Las distrofias musculares (DM) son entidades hereditarias que se caracterizan por debilidad muscular progresiva, pérdida de la masa muscular, hiporreflexia, fasciculaciones y discapacidad física variable. Son causadas por la mutación de uno de varios genes. De todas las DM, la distrofia muscular de Duchenne (DMD) es la más común, afecta a uno de cada 3,500 niños del sexo masculino; se caracteriza por debilidad y adelgazamiento muscular progresivos en la infancia, pérdida de la capacidad de caminar al final de la primera década de la vida y muerte a finales de la segunda década1-4. La distrofia muscular de Becker (DMB), es menos frecuente y se caracteriza por una progresión más lenta y mayor sobrevida de 5 ó 6 décadas5-7; 60% de los casos se heredan de manera recesiva ligada al sexo y las madres, que son portadoras de una mutación para la enfermedad, transmiten el gen mutado a la mitad de sus hijos que serán afectados y la mitad de las hijas serán portadoras sanas y transmitirán la mutación a la siguiente generación8. El gen mutado en los pacientes con DMD y DMB se localiza en el brazo corto del cromosoma X, en la banda Xp219 consta de 2,300 kb y 79 exones, se transcribe en un ARNm de aproximadamente 14 kb, que codifica para una proteína de 427 kD y 3,685 aminoácidos llamada distrofina. La ausencia o la disfunción de la distrofina causaría DMD/DMB y se ha propuesto que las fibras musculares de los afectados carecen de la interacción normal entre el sarcolema y la matriz extracelular; la interrupción de esta unión podría incrementar la fragilidad osmótica de las fibras musculares o alterar los mecanismos reguladores de los niveles de calcio, aumentar el flujo de iones Ca2+ en el músculo, hipótesis sustentada en los hallazgos histopatológicos. En ambos casos, habrá necrosis de las células musculares, lo que explicaría el cuadro clínico progresivo de la enfermedad10. Se ha descrito heterogeneidad mutacional en el gen de la distrofina que incluye deleciones, duplicaciones y mutaciones puntuales. La literatura mundial muestra que 66% de todas las mutaciones corresponden a deleciones o duplicaciones de uno o más exones en el gen11,12; las deleciones tienen una distribución no al azar a lo largo de la secuencia nucleotídica y se agrupan en dos regiones proclives o hot spots: 80% se concentran en la región de los exones 44 al 52 (cerca de la mitad afectan al exón 44)13; 20% restante comprenden los exones 1 al 1914-16. Debido al agrupamiento de las deleciones en dos puntos proclives, se ha establecido que el análisis de 18 de los 79 exones del gen de la distrofina, permite identificar 98% de todas las mutaciones del tipo deleción en el gen17-19; 33% de los casos, la mutación implica la alteración de un nucleótido único o de unos pocos nucleótidos16,20. La correlación genotipo-fenotipo se ha establecido mediante ejercicios meramente hipotéticos que relacionan el efecto de la mutación con el fenotipo de los pacientes. La hipótesis del corrimiento del marco de lectura traduccional (CMLT) propone que una vez ha ocurrido deleción, el gen resultante será el fruto de la reunión de los segmentos proximal presentándose dos alternativas: se preserva el marco de lectura (in frame); en este caso se conservan los dominios aminoterminal de unión con la actina y carboxiterminal de unión con los sarcoglicanos y distroglicanos de la membrana21-26 que permiten que se dé una proteína medianamente funcional con defectos en sus dominios internos, y causa una forma leve de la enfermedad o DMB, o contrariamente, la deleción crea un corrimiento del marco de lectura (out frame); en estos casos, aunque se conserva la región aminoterminal, el corrimiento en el marco de lectura distorsiona la proteína a tal grado que se produce una cadena polipeptídica diferente y no funcional, que impide que la proteína ejerza su función en la membrana del sarcolema y origina DMD con el cuadro clínico severo de la enfermedad21,22. Para evaluar los fenómenos out frame e in frame de las mutaciones que causan CMLT, se clasificaron los bordes de los exones como uno de tres tipos (1, 2 y 3), según la posición de los tripletes codificantes (Cuadro 1); una deleción que une dos exones con bordes del mismo tipo conserva el marco de lectura; por el contrario, una mutación que reúne dos exones con tipos de borde diferentes modifica el marco de lectura, conduciendo a la terminación temprana de la proteína o una proteína anormal, por una mutación del tipo missense o nonsense27. La mayoría de las mutaciones del tipo deleción, que afectan el marco de lectura traduccional, compromenten el dominio distal rod o remueven la región aminoterminal de la proteína; estas deleciones se han relacionado con el fenotipo severo de la enfermedad (DMD); en estos pacientes, por lo general, no se encuentra distrofina en la membrana del sarcolema28. En el presente trabajo se estableció en una cohorte de afectados colombianos con DMD o DMB, una correlación hipotética entre el genotipo-fenotipo al comparar el cuadro clínico de los afectados con los hallazgos moleculares y el análisis según la hipótesis del corrimiento del marco de lectura (CMLT)21. MATERIALES Y MÉTODOS Se analizaron 62 casos índice con DMD/DMB, remitidos por el Instituto Franklin Delano Roosevelt y la Asociación Colombiana de Distrofia Muscular (ACDM) provenientes de diferentes regiones de Colombia. Se registraron los datos personales y familiares (genealogía), así como los resultados de estudios de laboratorio (CPK, biopsia muscular y electromiografía); de acuerdo con la evolución clínica y los estudios paraclínicos se estableció el diagnóstico clínico del paciente, fuera DMD o DMB. Después de obtener el consentimiento informado de cada participante, se obtuvo por venopunción un tubo de 5 ml de sangre periférica total, de donde se extrajo el ADN genómico por el método de desalamiento23,29. Luego se hizo la amplificación por PCR múltiplex para 18 exones, con los iniciadores o primers diseñados por Chamberlain et al.30 y Restrepo et al.31 Las regiones amplificadas fueron el promotor (PM) y los exones 3, 4, 6, 8, 12, 13, 17, 19, 43, 44, 45, 47, 48, 50, 51, 52 y 60 (Gráfica 1). Los primers utilizados para la amplificación de los diferentes exones se sintetizaron en la casa comercial INVITROGEN en escala de síntesis de 100 nm, la solución de trabajo que se empleó correspondía a diluciones de 10 pm/ml. La mezcla de la reacción de PCR se llevó a un volumen final de 50 µl, mediante las siguientes concentraciones finales: amortiguador Taq polimerasa 1X (con 200 mM Tris-HCl pH 8.4; 500 mM KCl); 3.0 y 6.0 mM de MgCl2 (2.3 y 5.7 plex, respectivamente); 0.4 mM de cada dNTP (Promega); 5U de Taq ADN polimerasa (Promega) y 50 ng de ADN. Los cebadores correspondientes se mezclaron en sistemas de 7plex, 5plex, 3plex y 2plex en concentraciones de trabajo de 10 pml, la concentración final en la mezcla de reacción de PCR correspondió a 1.2 uM. Las muestras se corrieron en un termociclador PT100 MJ Research bajo las condiciones estandarizadas (Cuadro 2). Los productos amplificados se verificaron en un gel de agarosa al 2% (SIGMA) y confirmados en geles de poliacrilamida al 8%, que se tiñeron con bromuro de etidio; las bandas de amplificación se compararon con el patrón de peso molecular (puc18) (Gráficas 2, 3, 4 y 5). Los resultados se analizaron sobre transiluminador SIGMA y se obtuvo impresión fotográfica directa con cámara instantánea y rollos Polaroid en blanco y negro. RESULTADOS En 48 de las 62 familias analizadas el fenotipo del caso índice correspondió a DMD, mientras que el de los 14 (22.6%) restantes fue DMB. En los 48 casos de DMD se observaron deleciones en 14 (29%), mientras que hubo deleción en 5 (36%) de los 14 casos con diagnóstico DMB. Del total de 62 familias, 19 presentaron deleción, por lo que se puede afirmar que 31% de la población colombiana estudiada con DMD/DMB presentan deleciones en el gen de la distrofina (Cuadro 3). En la mayoría de los afectados hubo correlación entre el corrimiento (out frame) o no corrimiento (in frame) del marco de lectura traduccional y el cuadro clínico severo (DMD) y leve (DMB), respectivamente. Los afectados DM1, 2, 3 12, 18, 24, 39, 43, 46 y 65 mostraron deleciones del tipo out frame con fenotipos DMD. Por otra parte, los afectados con deleciones in frame, como los casos DM7, 29, 57, 58 y 79 presentaron DMB. La correlación del global del genotipo con el fenotipo fue 79%, pues los casos DM 9, 17, 44 y 55 presentaron el fenotipo severo pero tuvieron mutaciones in frame. En un informe preliminar de este estudio31, se presentaron los resultados analizados con dos sistemas múltiplex uno de 9 exones y otro de 5 exones, donde se analizaron 17 de las 27 familias por biología molecular, encontrando un porcentaje de deleciones detectables en 50% de las familias analizadas. DISCUSIÓN El hallazgo de una mutación en una secuencia específica de ADN, permite clasificar correctamente un diagnóstico genérico de DM en una de las distintas enfermedades específicas, p.e., DMD/DMB32. En el caso específico de DMD/DMB, además de confirmar el diagnóstico clínico, el hallazgo de mutaciones específicas del gen de la distrofina permite establecer en la mayoría de los casos, una correlación genotipo-fenotipo, ayuda a predecir el cuadro clínico del paciente, información que es esencial para el médico, el paciente y sus familiares21. Como se ha descrito antes, hay dos regiones proclives a deleciones en el gen de la distrofina19. En la presente cohorte se observaron deleciones del gen de la distrofina en 19 de 62 casos índice (31%). Al igual que en la literatura, en este trabajo se observa que 90% de las deleciones se presentaron en las dos regiones proclives: en los exones 1 al 19 se observaron 32%, mientras que 58% presentaron deleciones en los exones 44 al 53. Dos (10%) afectados mostraban deleciones que comprometían ambas regiones proclives, pero no escaparon al diagnóstico con el grupo de cebadores que amplifican los 18 exones propuestos en la literatura18. La frecuencia global de deleciones descritas en el presente trabajo es más baja que la media calculada (70%)33,34 y la de varias poblaciones individuales (Cuadro 4); sin embargo, es similar con los porcentajes informados en otros estudios de Latinoamérica que incluyen series publicadas en Argentina35 y Venezuela36; esto obliga a pensar que para la población colombiana el porcentaje esperado de duplicaciones, mutaciones puntuales e inversiones, debe ser más alto que el promedio descrito a nivel mundial16,37. En el Cuadro 3 se observa que los afectados con grandes deleciones que no interrumpen el marco de lectura, presentan la forma leve o DMB, de donde se concluye que la severidad de la enfermedad se determina más por el efecto intragénico que tiene la mutación sobre la proteína que por la extensión de la deleción. Del total de casos analizados se observó concordancia entre el genotipo y el fenotipo en 79% y al igual que en otras poblaciones, las mutaciones out frame correspondieron al fenotipo severo de la enfermedad29,35 (Cuadro 5). La excepciones a estas correlaciones pueden ser el resultado de alteraciones en los patrones de splicing, mutaciones in frame que generan codones prematuros de parada, restituciones del marco de lectura en mutaciones out frame en los exones del 3 al 7, entre otras38-41. En la población colombiana las excepciones a la correlación genotipo-fenotipo se dieron en pacientes con mutaciones in frame, cuyo fenotipo fue DMD, 3 de ellos con deleción del exón 19. El fenotipo se puede explicar si se tiene en cuenta que al interior de las 88 pb que forman parte el exón 19, se encuentra una secuencia de control de 52 pb, que actúa como señal en cis para la regulación del splicing del ARN heterogéneo nuclear, conduciendo el buen ensamblaje del spliceosoma, para que se dé un correcto ARN mensajero, que codifica para la distrofina42. Los elementos que actúan en cis, interactúan con elementos trans (como ribonucleoproteínas pequeñas nucleolares y otras proteínas auxiliares) y también dirigen el ensamblaje correcto del spliceosoma. El exón 19 además, tiene una región rica en purinas, que corresponde a una secuencia de reconocimiento de exones (ERS). Las ERS son necesarias para el splicing del exón inmediatamente anterior43 y promueven la inclusión (del exón 18) dentro del ARNm maduro por medio de una selección correcta de los sitios de corte. Esto sugiere que las deleciones que afectan los ERS, promueven una maduración errónea del ARNm, que a su vez conduce a un producto proteico alterado. Una isoforma conocida de la distrofina es la distrofina Kobbee producida por un gen que contiene una deleción de esa región de 52 pb dentro del exón 19, produciéndose un error en el splicing, aun cuando las secuencias 5’ y 3’ terminales del exón estén presentes43. El otro paciente (DMD)44 tuvo una deleción in frame que afectó los exones 48 al 51; sin embargo, el fenotipo de este paciente es severo (DMD), esto se puede atribuir a la generación de un codón de parada prematuro como consecuencia de la deleción, lo cual se puede corroborar mediante el análisis de la expresión del ARNm12,44. En conclusión, y porque este es el trabajo con mayor número de pacientes informados en Colombia, se puede afirmar que los afectados colombianos con DMD/DMB presentan un menor número de mutaciones por deleciones que la media universal y que inversamente, los casos debidos a otro tipo de mutaciones son más frecuentes aunque aún no se han estudiado en el país. Las deleciones se localizan en los dos puntos proclives en el gen de la distrofina, como se ha descrito en otros estudios y el juego de 18 primers propuesto en la literatura es adecuado para la identificación de deleciones en Colombia. Por último, la correlación entre el genotipo y el fenotipo fue posible en la mayor parte de los pacientes, bajo la hipótesis del CMLT y se puede utilizar como herramienta para establecer el diagnóstico y pronóstico de los afectados. AGRADECIMIENTOS Al doctor Alvaro Izquierdo y al Instituto Franklin Delano Roosevelt por el envío de pacientes y su confianza en los autores; a la Asociación Colombiana de Distrofia Muscular por su respaldo, confianza y colaboración; al Instituto de Ciencias Básicas de la Facultad de Medicina, Universidad del Rosario por el respaldo, tiempo y finaciación para el desarrollo de este trabajo. REFERENCIAS
© Copyright 2004 - Revista Colombia Médica The following images related to this document are available:Photo images[rc04040t2.jpg] [rc04040t5.jpg] [rc04040f4.jpg] [rc04040f5.jpg] [rc04040t3.jpg] [rc04040f1.jpg] [rc04040t1.jpg] [rc04040t4.jpg] [rc04040f2.jpg] [rc04040f3.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}