 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Revista Colombia Médica, Vol. 36, No. 4, Oct-Dec, 2005, pp. 266-270 Displasia campomélica. Descripción de un caso Case report: Campomelic dysplasia Erik Baltaxe, M.D.1, Fernando Suárez, M.D.2, Ignacio Zarante, M.D., M.Sc.3 1. Médico Rural, Instituto de Genética Humana, Facultad de
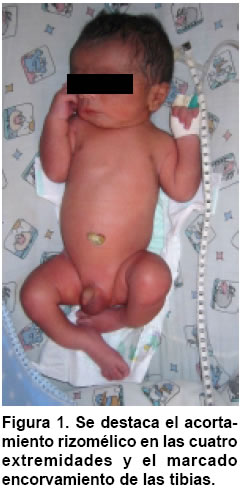
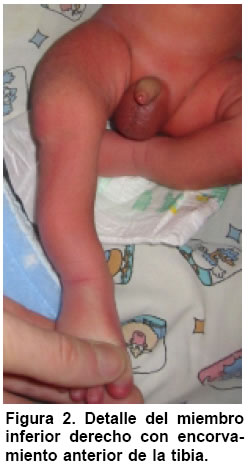
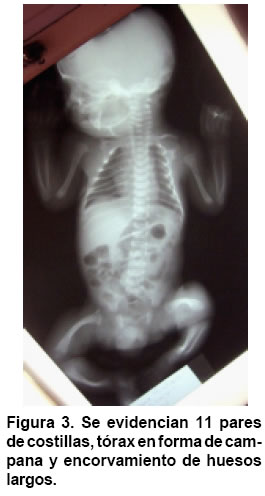
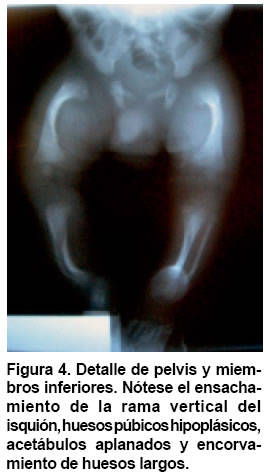
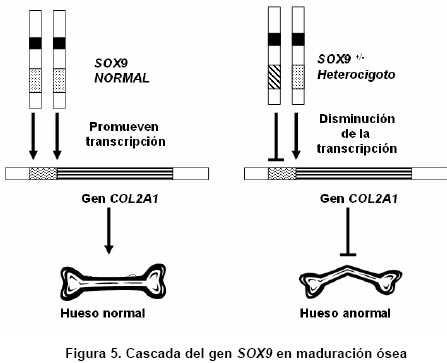
Medicina, Pontificia Universidad Javeriana, Bogotá, Colombia. e-mail:erik.baltaxe@javeriana.edu.co Recibido para publicación enero 26, 2005 Code Number: rc05063 RESUMEN La displasia campomélica es una alteración del desarrollo óseo que se presenta de forma austosómica dominante. Se caracteriza por el encorvamiento de los fémures y tibias, junto con otras alteraciones orofaciales, cardiopulmonares y neurológicas. El cariotipo puede mostrar sexo reverso. Las mutaciones del gen SOX9 son responsables en la mayoría de casos de estas alteraciones esqueléticas y genitales. Se presenta aquí un caso de displasia campomélica con compromiso óseo de miembros inferiores característico, identificado a través de ECLAMC (Estudio Colaborativo Latinoamericano de Malformaciones Congénitas) en Colombia y se hace una revisión de la fisiopatología molecular de la enfermedad. Palabras clave: Displasia campomélica; Displasia camptomélica; Malformaciones congénitas; Displasia esquelética; Reversión sexual; ECLAMC. SUMMARY Campomelic dysplasia is an alteration of bone development which is manifested as an autosomal dominant disease. It is characterized by femoral and tibial bowing, along with other items such as orofacial, cardiopulmonary and neurological alterations. The karyotyope results can show sex reversal. Mutations in the gene SOX9 are responsible in most of the cases for the skeletal and genital anomalies. A case of campomelic dysplasia with typical long bone bowing, identified in ECLAMC (Estudio Colaborativo Latinoamericano de Malformaciones Congénitas) is here presented and its molecular physiopathology is reviewed. Key words: Campomelic dysplasia; Camptomelic dysplasia; Congenital malformations; Skeletal dysplasia; Sex reversal; ECLAMC. Las displasias óseas agrupan un conjunto amplio y heterogéneo de alteraciones innatas del desarrollo del hueso y cartílago. La displasia campomélica, clasificada dentro de las osteocondrodisplasias letales, es una enfermedad caracterizada por el encorvamiento de los huesos largos, entre ellos la tibia y el fémur. Se puede asociar además con otras anomalías esqueléticas y extraesquléticas que comprometen la formación orofacial, genitourinaria, cardiopulmonar y neurológica. El estudio citogenético evidencia en algunos casos sexo reverso. Desde el punto de vista etiológico se han identificado las mutaciones del gen SOX9 como responsables en la mayoría de los casos. Su incidencia mundial se calcula en 1 de cada 200.000 nacimientos. En Colombia sólo se ha documentado este caso sobre una base poblacional de 45.000 nacimientos registrados por el ECLAMC (datos no publicados). PRESENTACIÓN DEL CASO El paciente fue visto en uno de los 11 hospitales vinculados al proyecto ECLAMC en Colombia. Previa firma de informe de consentimiento se diligenció el formato estándar sugerido por el ECLAMC y se revisó la historia clínica. Producto de primer embarazo de madre de 31 años natural y procedente de Bogotá, de ocupación abogada. Durante el embarazo cursó con infección urinaria manejada con ampicilina en el primer trimestre. Tuvo cuatro caídas desde su propia altura sin consecuencias aparentes. Consumió ácido fólico en el 1º y 2º trimestres. Antecedentes maternos farmacológicos: beclometasona tópica por rinitis alérgica. Paraclínicos: ecografías prenatales en número de 5. En la tercera, (con una edad gestacional de 20 semanas) mostró encorvamiento bilateral del fémur. Se practicó cesárea a las 35.5 semanas por ruptura prematura de membranas, y se obtuvo un producto de sexo masculino. El Apgar al minuto fue 8/10 dado por cianosis, 9/10 y 9/10 a los 5 y 10 min, respectivamente. El Silverman fue 1-2 y el Ballard de 37 semanas. Las medidas antropométricas fueron: peso: 2.280 g (P 25-50), talla: 44 cm ( < P 10), perímetro cefálico: 32 cm (P 25), distancia intermamilar: 7 cm (P 3-50), mano total: 6 cm (P 50) y dedo medio: 2.5 cm (P 50). Al examen físico presentaba fontanelas amplias, sutura coronal abierta, ictericia leve. Cardiopulmonar, tórax y abdomen normales. Las extremidades mostraban acortamiento rizomélico en miembros superiores; encorvamiento en fémur y tibia innegables clínicamente(Figuras 1 y 2) Examen neurológico normal. Requirió Unidad de Recién Nacidos por prematurez, taquipnea transitoria del recién nacido y sospecha de infección neonatal secundaria a ruptura prematura de membranas (14 horas). Duró hospitalizado 8 días con mejoría del cuadro respiratorio e infeccioso, y egresó estable clínicamente. Paraclínicos. Estudio radiológico: Tórax AP y lateral, cuerpo entero y miembros inferiores. Deformidad campomélica solamente de los huesos largos de miembros infe-riores, con alteraciones en la forma del tórax, el número de costillas y los huesos pélvicos (Figuras 3 y 4). FTA ABS. No reactivo. Serología (RPR): No reactivo. Química sanguínea al nacimiento: BUN: 3.5 mg/dl; creatinina: 0.58 (0.7-1.5) mg/dl; calcio: 10.13 (8.4-10.2) mg/dl; fósforo: 6.06 (2.5-4.5) mg/dl; fosfatasa alcalina 375 (38-126) U/l; bilirrubina total 8.9 mmol/l; bilirrubina directa 0.16 mmol/l. Química sanguínea de control. Calcio: 10.01 (8.4- 10.2) mg/dl; fosfatasa alcalina: 429 (38-126) U/l; fósforo: 6.44 (4-7) mg/dl. Cariotipo. 46, XY sin alteraciones cromosómicas estructurales. En el control a los 5 meses se encontró un paciente asintomático, con evolución favorable y sin progresión clínica del encorvamiento femoral. Las radiografías de control de miembros inferiores sugieren como diagnóstico diferencial hipofosfatasia, sin descartar otras etiologías. DISCUSIÓN Además de sus alteraciones óseas características la displasia campomélica se puede asociar también con otras anomalías esqueléticas como macrocranea, cara aplanada, cifoescoliosis, talipes equinovaro, hipoplasia escapular y costillas delgadas. Igualmente puede haber otras alteraciones extraesquléticas como paladar hendido, hidronefrosis, hipotonía y dificultad respiratoria que en muchos casos es la causa de muerte. Asimismo se han descrito casos con malformaciones cardíacas, retraso mental y sordera (1). Radiológicamente se consideran las siguientes características: mandíbula hipoplásica, vértebras cervicales hipoplásicas, escápula hipoplásica, tórax pequeño en forma de campana con once pares de costillas, pedículos vertebrales hipoplásicos o no mineralizados, hueso ilíaco con rama vertical delgada, hueso púbico hipoplásico, luxación de cadera con acetábulo aplanado, tibia y fémur encorvados, osificación retardada de las epífisis distales del fémur y proximales de la tibia y esternón, cabezas radiales luxadas, primeros metacarpianos cortos y falanges medias cortas del segundo al quinto dedos (2). Esta displasia esquelética es infrecuente y se calcula su incidencia en 1 caso por cada 200.000 nacimientos (1). Casi siempre es una enfermedad letal, pero se estima que 10% de los afectados sobreviven. Estos pacientes desarrollan cifoescoliosis severas, talipes equinovaro, luxación o subluxación congénita de la cadera, luxación de las cabezas radiales con limitación en la supinación, retardo en el desarrollo motor (en especial motor grueso), déficit auditivo y caries dentales (3). En este caso se deben considerar los diagnósticos dife-renciales que se mencionan en el Cuadro 1(4-10). En algunos recién nacidos que son fenotípicamente femeninos se puede encontrar un cariotipo 46, XY (sexo reverso). El espectro comprende desde un fenotipo femenino hasta genitales ambiguos (1,2). La causa de esta alteración esquelética se encuentra en las mutaciones del gen SOX9 que se localiza en el cromosoma 17. Este gen es responsable de la diferenciación sexual y de la maduración de los condrocitos (1,11). El gen codifica una proteína de 509 aminoácidos que hace parte de la familia de los factores de transcripción tipo SRY-SOX12. Desde un punto de vista fisiopatológico, la mayoría de mutaciones presentes en la displasia campomélica afectan el sitio de unión de la proteína SOX9 al ADN. Recientemente se han descrito mutaciones que afectan la dimerización de esta proteína, e impide que realice su función una vez unida a la cadena de ADN (11,13). El gen SOX9 codifica para el factor de transcripción que hace que los condrocitos produzcan el colágeno tipo II normal del hueso, a través de la activación del gen COL2A1. Si alguna de las copias del gen SOX9 está mutada no se activará la transcripción del gen COL2A1 de manera que se producirá un colágeno anormal (Figura 5). Este mecanismo se llama haploinsuficiencia porque se comporta como si el gen SOX9 estuviera en una sola dosis (haploide) (14). Los condrocitos interpretan la baja acción de SOX9 como el estímulo para pasar a la fase de hipertrofia y mineralización ósea. Esto lleva consigo a la mineralización prematura de los núcleos de crecimiento epifisiarios y por tanto a la detención del crecimiento (12,14). El encorvamiento de los huesos largos se puede explicar por la tracción que generan los músculos adyacentes sobre un hueso debilitado por la ausencia de colágeno normal (14). Lo anterior podría sugerir el mecanismo autosómico dominante para la herencia de esta entidad. En cuanto a la diferenciación sexual, las interacciones son más complejas y no se han dilucidado por completo. Se sabe, sin embargo, que el gen SOX9 ejerce un efecto que depende de la dosis en el desarrollo y supervivencia de las células de Sertoli. Además SOX9 se une al gen que activa la hormona antimülleriana (AMH), de modo que cuando la proteína SOX9 se altera, hay ausencia total de transcripción en AMH lo que lleva a la persistencia de los conductos de Müller en ratones transgénicos (12). Esto explica la presencia de un fenotipo femenino con un cariotipo masculino XY. Se han propuesto varios mecanismos fisiopatológicos para explicar el hallazgo conjunto de encorvamiento más sexo reverso o cada característica por separado.
El asesoramiento genético se debe enfocar hacia un mecanismo de herencia autosómico dominante donde en la mayoría de las ocasiones se trata de una mutación de novo. Se ha descrito mosaicismo gonadal o somático en los padres de pacientes afectados, lo que genera un riesgo de recurrencia empírico de 2% a 5% (16). Esta entidad es uno de los mejores ejemplos de “fisiopatología molecular” aplicada a la genética clínica y muestra cuál debe ser el objetivo de un grupo de estudio multidisciplinario que integre de modo adecuado los conocimientos de las ciencias básicas en la comprensión de la enfermedad. AGRADECIMIENTOS Al doctor Roberto Mendoza, Baylor College of Medicine, Houston, Texas por sus comentarios sobre el caso. Al doctor Fernando Novoa, radiólogo del Hospital Universitario San Ignacio por la interpretación de las imágenes radiológicas. Este trabajo se llevó a cabo con datos del Estudio Colaborativo Latinoamericano de Malformaciones Congénitas (ECLAMC). REFERENCIAS
© Copyright 2005 - Revista Colombia Médica The following images related to this document are available:Photo images[rc05063f2.jpg] [rc05063t1.jpg] [rc05063f1.jpg] [rc05063f5.jpg] [rc05063f3.jpg] [rc05063f4.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}