 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Revista Colombia Médica, Vol. 37, No. 2, Apr./Jun. 2006, pp. 114-120 A comparative, cross-over, double blind, randomized study for bioequivalence assessment between two formulations of valsartan capsules vs. tablets Estudio comparativo, cruzado, doble ciego, al azar para determinar la bioequivalencia entre dos formulaciones de valsartán en tabletas y cápsulas Milena Pérez, Quim.1, William Cárdenas, M.D.2, Gloria Ramírez, Quim.3, Mauricio Pérez, Estat.4, Piedad Restrepo, Farmacol.5 1. Director of the
Biopharmaceutical Laboratory, CIDEIM, Cali, Colombia. e-mail:
milena_perez@cideim.org.co Recibido para publicación septiembre 19, 2005 Aceptado para publicación marzo 15, 2006 Code Number: rc06018 SUMMARY Introduction: Bioequivalence or compared equivalence studies are used
to demonstrate claims that the new product, named generic product,
will have the same bioavailability as the reference product,
named brand product. If two products are bioequivalent it means that
they would expect to have the same efficacy and security.
Bioequivalence is established by the statistical estimation of
significant differences or not in the pharmacokinetics parameters of
area under the curve (AUC) and maximum concentration (Cmax). In this
case, bioequivalence will be evaluated and the bioavailability of
Valsartan will be compared. Valsartan is an agent antihypertensive
and specific angiotensin II antagonist acting on the AT1 receptor
subtype. Key words: Valsartan; Bioavailability; Bioequivalence. RESUMEN Introducción: Los estudios de bioequivalencia o equivalencia
comparada se realizan para demostrar que el producto en estudio,
conocido como producto genérico, tiene la misma
biodisponibilidad del producto de referencia, también conocido
como producto innovador o de marca. Si los dos productos son
bioequivalentes, se espera que tengan las mismas
características de seguridad y eficacia. La bioequivalencia es
establecida por la estimación estadística de
diferencias significativas o no en los parámetros
farmacocinéticos de área bajo la curva (ABC) y
concentración máxima (Cmáx). En este caso, se
evaluará y se comparará la biodisponibilidad de
valsartán, un agente antihipertensivo inhibidor
específico del receptor de angiotensina II subtipo AT1, en las
membranas celulares del músculo liso vascular. Palabras clave: Valsartán; Biodisponibilidad; Bioequivalencia. The principle active valsartan is a nonpeptide, orally active and specific angiotensin II antagonist acting on the AT1 receptor subtype. Valsartan is a white to practically white fine powder; it is soluble in ethanol and methanol and slightly soluble in water. Valsartan is chemically described as N-(1-oxopentyl)-N-{[2’-(1H-tetrazol-5-y1)[1,1’-biphenyl]-4-y]methyl}-Lvaline. Its empirical formula is C24H29N5O3, its molecular weight is 435.5, and its structural formula is (1):
Angiotensin II is formed from angiotensin in a reaction catalyzed by angiotensin-converting enzyme (ACE, kininase II). Angiotensin is the principal pressor agent of the renin-angiotensin system, with effects that include vasoconstriction, stimulation of synthesis and release of aldosterone, cardiac stimulation, and renal reabsoption of sodium. Valsartan blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the AT1 receptor in many tissues, such as vascular smooth muscle and the adrenal gland. Its action is therefore independent of the pathways for angiotensin II synthesis (2). Valsartan was the second nonpeptide At-II type 1-receptor antagonist available for the treatment of hypertension. Valsartan is rapidly absorbed from the gastrointestinal tract after oral administration and can be administered without regard to food intake. The peak effect of valsartan is evident in two to four hours; the bioavailability is 25%. Valsartan has a half-life of six to nine hours and demonstrates antihypertensive effects for approximately 24 hours. Less than 10% of an orally admi-nistered dose of valsartan undergoes biotransformation in the liver; the enzymes responsible for its metabolism are known, and no active metabolites have been identified. Elimination occurs primarily in the bile (86%) and to lesser extent via the kidneys (13%), largely as unchanged drug (3). The tests of bioavailability are based on the application of concepts with scientific evidence in biological and pharmaceutical sciences to demonstrate the efficiency of drugs in giving the substances with pharmacologic activity that contain, in the waited for amount and the time in agreement with the design, development and quality declared by the manufacturer. The tests of bioavailability are component of the previous tests to the approval like condition to obtain the sanitary registry of any new drug in the market and are made during the clinical studies of the products that contain an active principle never before used or indicated in certain pathology or a new presentation as pharmaceutical forms for administration by routes different from the intravenous. The characterization of the product as far as its bioavailability, this is, as far as the amount and the speed of liberation and arrival of the active principle to the organism, it bases the estimation of the effectiveness properties, security and quality of the same one as guide who allows to value the efficiency of the developed product, it facilitates the comparison of this with other similar products, today more known like generic drugs, elaborated under different conditions by types of materials, procedures, productive circumstances or different surroundings. The studies to establish bioequivalence (BE) are carried out to identify significant statistical differences during the behavior in vivo of pharmaceutical equivalent products, that they have the same amount of active principle; they appear in the same form of dosage and fulfill the requirements of the pharmacopoeias officially accepted for the check tests of quality to finished products (4). For many products, the comparison is based on the evaluation of indicative parameters of the bioavailability, as they are the area under the curve elaborated with the changes of concentration in a biological fluid, detected during a period of time, and the maximum concentration obtained for each product. The studies are applied to drugs that are administered by non-parenteral routes, where the innovator or product with which the investigations of effectiveness and security for the drug were carried out, it is denominate of «reference» is considered (R), and the new product to commercialize itself denominates the product of «test» (T) (5). The demonstration of bioequivalence between products, by means of an suitable design of investigation, it allows that the sanitary authority admits the declaration of substitution and interchangeability among them, during the dispensation or therapeutic use (6). The aim of this study was to evaluate the relative bioavaibility of two tablet formulations both containing 80 mg valsartan. MATERIALS AND METHODS Volunteers. The study was made in fourteen male healthy volunteers aged 18-29 years (weight 50-84 kg). The Ethic Institutional Human Investigations Committee of CIDEIM reviewed and approved in the corresponding to fulfillment of the requirements of the World Health Organization and the Colombian norms, the content of the protocol of this study and the consent informed which was obtained after the nature and possible consequences of the studies were explained. Chemicals. Valsartan was obtained from USP. Losartan used as an internal standard was obtained from Chemo. Acetonitrile (Fisher Scientific) and water was HPLC grade. Phosphoric acid (Fisher Scientific), methanol (Panreac), and potassium monobasic phosphate (Merck), were analytical grade. Instrumentation and chromatography. Chromatography was performed with a high-performance liquid chromatograph LaChrom Elite (Merck-Hitachi) and an UV detector with diodes array, Chromolith® Performance RP-18e 100-4.6 mm, 2 µm. Merck, Data station with EZChrom Elite 3.1.3 software Merck, C8 cartridges Sep-Vac (100 mg, 1 ml) de Waters. Ohaus Adventurer analytic balance, Vortex shaker Fisher Scientific, Magnetic shaker Multistation IKA, Ultrasonic bath Fisher Scientific, Water bath DIES, Vaccum system Labconco, Vaccum pump Precision, Ultra Pure Water Productor Simplicity Millipore, Centrifuge Sorvall RC-5B, Freezer -70ºC Revco, pH meter WTW Inolab pH 740, solid-phase extraction equipment Supelco and air compressor Atlas Copco. DESIGN AND CONDUCTION OF THE STUDY The study was designed for to evaluate the bioavailability of the pharmaceutical products identified as valsartan A, tablets capsules 80 mg, and valsartan B tablets 80 mg, with base in the pharmacokinetics measures of amount and velocity (in terms of required time), where upon the valsartan reaches the sanguineous circulation after being administered by oral route to fifteen volunteers, in dose of 320 mg of each product, in at random assigned and separated periods of treatment to each other by an interval of fifteen days. CHEMICAL EQUIVALENCE The samples of products administered to the volunteers were evaluated previously for the fulfillment of the physicochemical properties. The results for both samples allowed to conclude conformity with the applied methods for: organoleptics characters, dimensions, weight average, identification and assay of the active principle, test of dissolution and uniformity of content. The results of the evaluation tests verified the fulfillment of the specifications of the manufacturer for both products and therefore chemical equivalence between them. EXPERIMENTAL DESIGN Study was made of bioequivalence with base in the compared bioavailability of 320 mg single doses of valsartan in tablets and capsules administered by oral route, in at random crossed design of two routes for the product of test valsartan A, 80 mg capsules, vs. the reference standard valsartan B, 80 mg tablets, and its dispensation was in blind for the researchers and volunteers subjects. The products were administered by random allocation to fifteen male healthy volunteers aged 19-28 years (weight 60-79 kg). The study was performed according to a conventional cross-over experimental design, not replicated, of two formulations, in two periods and two sequences, and with random assignment of the subjects to each treatment, in order to determine the average values (in arithmetic and logarithmic scales), and to establish the differences and ratios, between these average values for the two products (R and T). The calculations were based on the data generated for the valsartan analyte in relation to: area under the concentration curve in time (AUC) between zero and t and zero and infinite; maximum concentration achieved (Cmax) and time for maximum concentration. The products were coded by the Biometry Unit in CIDEIM as B 80 mg tablets and A 80 mg capsules to ensure that all participants in the trial (researchers and subjects alike) remained blind as to which product was administered at which period to which subject. The sequences of administration were randomly selected and the identity of the volunteers was protected by using a numeric code instead of a name in all the necessary paperwork. CLINICAL PROCEDURES On the day of the study at 7:00 a.m. to each subject was putt in a heparinized catheter on the an arm vein, then, 15 ml of blood were extracted from each subject and were labeled as «sample zero»; afterwards, each subject received one dose corresponding to 320 mg of each product according to the random order assigned, with 240 ml of dextrose solution 20%, and starting at that moment, 13 more samples (15 ml each) were extracted at the times 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10 and 12 hours. Each blood sample obtained was labeled before sending it to the clinical laboratory for the plasma separation and prior to the physico-chemical analysis. The second assay period was separated from the first one by 8 days, in order to assure sufficient elimination of the product already administered. ANALYTICAL PROCEDURE The analytic technique was standardized, validated and applied to the plasma samples from healthy volunteers according to the standardized operating procedures code SOP10033, SOP10044 and SOP10045. The procedure and technical information related to it are elaborated based on the industry guides published by the FDA of the United States, among them the ones described in regulations 21CFR part 58 and 21CFR 320.29, which have details related to: specificity and minimum limit of quantification. Calibration curve and linearity. Analyzed in a matrix with internal standard, with concentrations in the range of valsartan 50-20000 mg/ml and standard addition of valsartan 2500 mg/ml. Correlation coefficient was 0.9927. Precision, accuracy and recuperation.The precision was determined 3 times for every concentration and for 20 concentrations in the range. Specificity was determined with a precision of 20% and accuracy between 80% and 120%. Recuperation was determined by comparison between the measurements of the analyte in the sample and in a standard preparation and it’s was 90%. The analytical method applied to the determination of the content of valsartan in plasma samples was developed based on a methods referenced in the bibliography (1,2). The applied technique was high performance liquid chromatography (HPLC). The equipment used was a LaChrom Elite® liquid chromatographer with a diodes array detector. The analyte was evaluated according to the standardized and validated procedure, with ultraviolet radiation at 265 nm, solid phase extraction through the use of C8 Sep-Pak of Waters cartridges and a Chromolith® C18 100 x 4.6 mm column with 2 mm of particle size; losartan was used as the internal standard and the mobile phase comprised a mixture of solvents (acetonitrile-buffer potassium phosphate (45:55) adjusted to pH 2.7 with acetic acetic glacial); the separation time of the compounds was 10 minutes. KINETIC PARAMETERS Peak plasma concentration (Cmax) and time to peak (Tmax) were obtained of the plasma concentration-time curve of valsartan. Area under the plasma concentration curve AUCo-twas calculated by the trapezoidal rule and AUCt-inf was calculated by last concentration estimated and slope of the phase of elimination. STATISTICAL EVALUATION The bioequivalence was determinate for the application of statistic calculations with a confidence interval of 95% for the difference between the mean values obtained for the assayed product valsartan A or T (test product) and valsartan B or R (reference product), in relation to variations of the area under the concentration-time curve (AUC) and of the maximum concentration (Cmax) for the studied formulations, after the logarithmic transformation of the data. The antilogarithmic values of the confidence limits will constitute the interval of 95% for the ratio of the geometric averages between products A y B, with the acceptance of a confidence interval of at least 80% and no more than 125%, through of the applying of the two applications of test of unilateral hypotheses, defined like: Ho: ratio between logarithmic averages A/B less or equal to log(0.8) vs. Ha: ratio between logarithmic averages A/B greater than log(0.8); Ho: ratio between logarithmic averages less or equal A/B to log(1.25) vs. Ha: ratio between logarithmic averages A/B greater than log(1.25) in order to reject the null hypotheses and to establish the bioequivalence between products, defined like the difference between the averages included in the interval from 0.8 to 1.25 (80% to 125%), with 5% of significance. The following information required for the pharmacokinetic analyses will be described:
In addition, the statistical information for ABC0-t, ABC0-∞ and Cmax will be obtained, referring to:
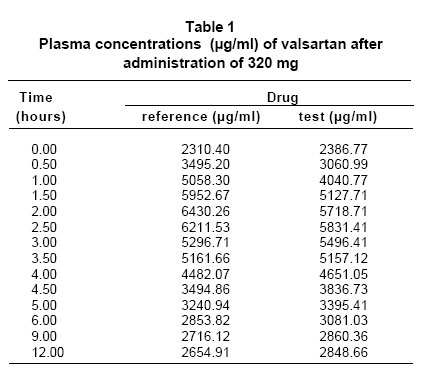
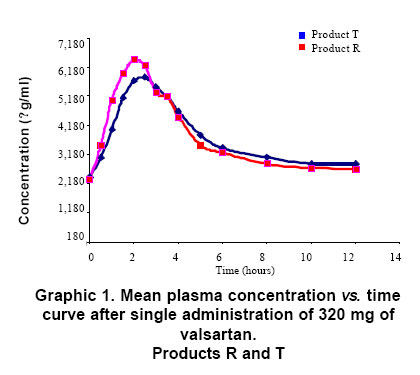
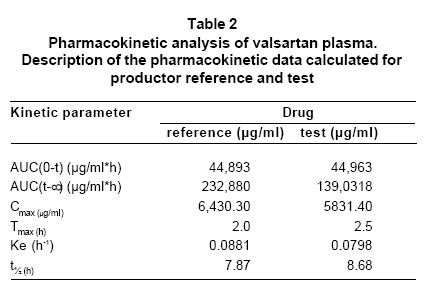
RESULTS The comparison of bioavailability, of the product «test» (T, code To for this study) and the product «reference» (R, code B for this study), was made with base in the values of primary data of valsartan concentration of in the time, and of the secondary data for the pharmacokinetics of area under the curve of plasmatic concentrations between time zero and t. There were statistically no significant differences between two products (A and B), for transformed variables AUC (level of significance 0.874), Cmax (level of significance 0.067) and Tmax (level of significance 0.688) (Table 1, Graphic 1). The comparison of the values of the pharmacokinetic parameters or secondary data, calculated with base in the experimental data for AUC and concentration, by the application of the non-compartmental model and the ratio between the values obtained for both products, they show the bioequivalence between products both, with which the exposed results are corroborated already (Table 2). Through of the test of Shapiro-Wilk the normality of the two variables is confirmed. Test t of twin Student is used. Were not differences between the concentration average of products A and B (value p=0.487). The differences of the values average of Cmax and AUC, in the primary data and with logarithmic transformation fulfill the criterion of values of significance over 0.05 what allows to establish the significant non difference between products A and B with a level of 95% confidence. In the tests of hypothesis for the ratio of the averages of the primary data and with logarithmic transformation of AUC and Cmax the null hypothesis was rejected (Ho), in the limit inferior of the ratio between the averages of A/B less or equal to 80% or 0.8 in data primary or transformed respectively. And was approved the null hypothesis in the superior limit of the ratio between the averages of A/B less or equal 120% or 1.25 in data primary or transformed respectively. The previous results allow to establish the significant non difference between products A and B as far as the valsartan amount of that reaches the sanguineous circulation and the level of maximum concentration, during twelve hours after administering an 320 mg dose by oral route to healthy subjects, with a level of confidence of 95% and greater significance of 0.05. DISCUSSION The analytical results and the statistical analysis of such base the conclusion of bioequivalence between products A and B, for the speed and the reached amount of valsartan in the organism, after being administered in single doses to 15 healthy volunteers subjects, according to the experimental design crossed allocation at random of the treatments and the periods. The application of the criteria accepted in literature and the referenced regulation in this study, it demonstrated in the comparison of products A and B, that the ratio between the area under the curve of concentration valsartan in the time, and the ratio between the maximum concentration obtained with each product, were within the acceptance limits, as much for the experimental data as for the resultants of its logarithmic transformation. In agreement with the results of the analytical determination of the active principle valsartan in the plasma in 15 volunteers subjects, the results of the statistical and pharmacokinetic analysis for the comparison, the referenced guides and regulations that describe to the limits of acceptance of bioequivalence between drugs and the fulfillment of the objectives for this study, concludes the bioequivalence between products A or product in test and product B, or of reference. CONCLUSION The design of this study and the application of the protocols chosen enabled the demonstration of bioequivalence between the products Valsartan MK®/valsartan 80 mg capsules from Tecnoquímicas Laboratories and Diovan®/valsartan 80 mg tablets from Novartis Laboratories. ACKNOWLEDGMENTS The authors thank the Colombian Institute for the Development of Science and Technology (COLCIENCIAS) for financial support of the project and the company Tecnoquímicas S.A. for financial support of the bioequivalence study and for providing quality standards of valsartan and losartan. The authors thank to doctor Omar Velásquez from Ecoquímica for the scientific advisory and technical support in the validation of the analytical method for to determinate valsartan in human plasma by HPLC/UV. REFERENCES
© Copyright 2006 - Revista Colombia Médica The following images related to this document are available:Photo images[rc06018t2.jpg] [rc06018f1.jpg] [rc06018t1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}