|
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Revista Colombia Médica, Vol. 37, No. 4 , Oct./Dec. 2006, pp. 315-318 El síndrome de Kallmann: A propósito de un caso Kallmann’s syndrome: A propos of a case William Jubiz, M.D.1, Eduardo Antonio Cruz, M.D.2 1. Profesor, Medicina
Interna y Ginecología Endocrina, Facultad de
Salud, Universidad Libre. Director, Centro de
Endocrinología, Metabolismo y Diabetes, Cali,
Colombia. e-mail: jubizwill@uniweb.net.co Recibido para
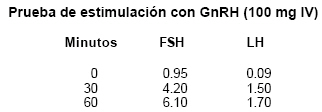
publicación octubre 3, 2005 Code Number: rc06061 RESUMEN El síndrome de Kallmann es un tipo de hipogonadismo hipogonadotrópico que puede afectar a hombres y mujeres; se caracteriza por hábito eunucoide, deficiente desarrollo sexual y anosmia por desarrollo defectuoso de los bulbos olfatorios. También puede ocurrir paladar hendido, sordera, convulsiones, cuarto metacarpiano corto, anomalías cardíacas y ginecomastia. La transmisión genética puede ser autosómica dominante, autosómica recesiva o ligada al cromosoma X. En esta última se presentan mutaciones o deleciones del gen KAL, localizado en Xp 22.3, el cual codifica la síntesis de anosmina-1, una proteína asociada con funciones de adherencia celular y actividad antiproteasa. Las concentraciones de la testosterona sérica así como la de la hormona folículo-estimulante (FSH) y luteinizante (LH), están disminuidas pero hay respuesta a la administración de la hormona liberadora de gonadotropinas (GnRH). La infertilidad se trata con una combinación de gonadotropina coriónica (hCG) y gonadotropina menopáusica humana (hMG). La deficiencia androgénica se corrige con testosterona cuyas formas más útiles son el enantato (Testoviron®) y undecanoato (Nebido®) parenterales, los parches (Androderm®, Testoderm®) y los geles (Androgel®, Testim®). Existe un preparado de testosterona de absorción bucal (Striant SR®), el cual parece ser efectivo y conveniente. Se presenta un paciente con síndrome de Kallmann quien consultó a los 18 años por retardo del desarrollo sexual. No podía oler. Tenía testículos y pene pequeño, eunucoidismo, testosterona baja con FSH y LH bajas y una respuesta subnormal a GnRH. Respondió a la administración de testosterona con aparición de vello púbico y axilar, aumento del tamaño del pene y engrosamiento de la voz. Palabras clave: Síndrome de Kallmann; Hipogonadismo; Testosterona; Gonadotropinas. SUMMARY Kallmann’s syndrome is a type of hypogonadotropic hypogonadism which affects males and females and is characterized by eunuchoidal habitus, lack of sexual development, and anosmia, caused by a defective development of the olfactory bulbs. Cleft palate, deafness, seizures, short fourth metacarpal bones, cardiac abnormalities and gynecomastia may also occur. The mode of transmission can be autosomal dominant, autosomal recessive or X-linked. The latter is caused by mutations or deletions of the KAL gene which encodes the synthesis of anosmin-1, a protein associated with cellular adherence and antiprotease activity. The concentrations of testosterone, follicle stimulating hormone (FSH) and luteinizing hormone (LH) in serum is decreased, but they respond to the administration of the gonadotropin releasing hormone (GnRH). Infertility is treated with a combination of human chorionic gonadotropin (hCG) and human menopausal gonadotropins (hMG). Androgen deficiency is corrected with testosterone in the form of parenteral enanthate (Testoviron depot®) or undecanoate (Nebido®), patches (Androderm®, Testoderm®) or gels (Androgel®, Testim®). Sriant SR® is absorbed through the oral mucosa and it appears to be effective and convenient. An 18 year-old male who consulted for sexual retardation is presented. He could not smell. Testes and penis were small and he had an eunuchoidal habitus. Serum testosterone, follicle stimulating (FSH) and luteinizing (LH) hormones were decreased with a subnormal response to gonadotropin-releasing hormone (GnRH). He responded to testosterone therapy with the development of axillary and pubic hair, increased penis size, and deepening of the voice. Keywords: Kallmann’s syndrome; Hypogonadism; Testosterone; Gonadotropins. La consulta y evaluación de un paciente con síndrome de Kallmann motivó la revisión del tema, en la cual se incluyó el cuadro clínico, genética, diagnóstico y tratamiento. El síndrome descrito originalmente por Kallmann et al.1,2 se caracteriza por deficiencia aislada de gonadotropinas y anosmia, debidas a un desarrollo defectuoso de los bulbos olfatorios. Son parte del síndrome paladar y labios hendidos, sordera, convulsiones, cuartos metacarpianos cortos y anomalías cardíacas3. La incidencia del síndrome es de 1 en 10,000 nacimientos de varones. Tanto hombres como mujeres pueden ser afectados. Los hombres se presentan como eunucos prepuberales. Por lo general no hay desarrollo puberal y las proporciones esqueléticas son eunucoides con disminución en la relación de la distancia vértice-pubis sobre la distancia del pubis al piso. La envergadura (distancia de dedo medio a dedo medio de la mano con los brazos abiertos), tiene más de 6 cm que la talla. Estas proporciones se deben a una falla de la fusión epifisaria que permite continuación en el crecimiento de los huesos largos. El vello púbico y la barba son escasos o no existen. La anosmia es muy característica4,5. El pene es pequeño y los testículos son prepuberales pero blandos y no duros como ocurre en el síndrome de Klinefelter. Puede haber ginecomastia por aromatización periférica de los andrógenos suprarrenales. No hay retardo mental4,5. GENÉTICA Con más frecuencia el síndrome de Kallmann se asocia con una herencia autosómica dominante2. La anosmia ha permitido establecer una transmisión de padre a hijo6. También hay informes de transmisión autosómica recesiva o ligada al cromosoma X7. Esta última se debe a mutaciones y deleciones en el gen KAL que se localiza en Xp22, 38-10. El gen consiste de 14 exones y produce una proteína de 680 aminoácidos y 76 kda llamada anosmina-1. La proteína se asocia con funciones de adherencia celular y actividad anti-proteasa. También hay informes de mutaciones del gen del receptor del factor de crecimiento de los fibroblastos (FGFR-1) lo cual conduce a agenesia de las neuronas olfatorias y secretoras de la hormona liberadora de gonadotropina, GnRH11,13. LABORATORIO Los niveles basales de gonadotropinas (FSH, hormona folículo estimulante) y LH, hormona luteinizante), están bajos con una pobre respuesta inicial a la administración de GnRH pero con respuesta normal cuando la hormona se inyecta en pulsos en forma repetida14-16. Esto indica que el defecto radica en la secreción hipotalámica de GnRH. La anatomía de la hipófisis es normal y la secreción de las otras hormonas hipofisarias es normal. La concentración plasmática de testosterona está baja y la de prolactina es normal4,5. TRATAMIENTO El manejo de los pacientes con el síndrome de Kallmann tiene dos objetivos: mejorar la fertilidad y suministrar terapia androgénica. La fertilidad se puede restaurar de dos maneras. La primera consiste en la administración de gonadotropina coriónica humana (hCG) seguida de FSH como gonadotropina menopáusica humana (hMGs). El tratamiento previo con andrógenos no interfiere con la respuesta17. La dosis inicial de hCG es de 1,000 a 2,000 UI intramusculares tres veces por semana que estimula las células de Leydig y aumenta la concentración de la testosterona intratesticular. De las semanas 8 a 12 de tratamiento, cuando las concentraciones plasmáticas de testosterona hayan aumentado, se inicia hMG, 75 UI intramusculares tres veces por semana, mientras se continúa la hCG. Una vez que se inicie la espermatogénesis se disminuye la dosis de hMG a 12-25 UI tres veces por semana. En algunos pacientes se mantiene la espermatogénesis sin hMG. El segundo enfoque consiste en la administración de pulsos subcutáneos de GnRH cada 90 a 120 minutos. La dosis por pulsos es de 1 a 5 mg, según lo que se necesite para obtener una concentración normal de testosterona sérica. La espermatogénesis ocurre generalmente a los dos o tres meses14,16. Cuando el síndrome de Kallmann se presenta en mujeres, la fertilidad también se puede restaurar por medio de la administración de GnRH. Terapia androgénica. La terapia androgénica produce efectos benéficos de diferente índole. El comportamiento sexual mejora con aumento de la libido y la erección. Además, los pacientes experimentan un aumento de la energía, sensación de bienestar, actividad física, motivación e iniciativa, y una mejoría de la memoria y el sueño18,19; la testosterona aumenta la masa muscular, disminuye la grasa, aumenta la densidad del mineral óseo y estimula la eritropoyesis. La testosterona está disponible en diferentes formas para administración por varias vías. Las formas más utilizadas son el enantato y cipionato de testosterona. La dosis inicial es de 200 mg intramusculares cada dos semanas, la concentración plasmática de testosterona aumenta rápidamente en uno o dos días y se mantiene dentro de los límites normales por dos semanas. El preparado parenteral más novedoso es el undecanoato de testosterona (Nebido®), que se inyecta por vía intramuscular en dosis de 1,000 mg cada 12 a 15 semanas. Con este medicamento la concentración plasmática de testosterona es estable20. La vía transdérmica se ha popularizado en los últimos años. El compuesto hidroalcohólico de testosterona al 1% (Androgel®) se aplica diariamente en dosis de 50, 75 ó 100 mg. Los niveles sanguíneos de testosterona se normalizan y mantienen el ritmo circadiano normal de la hormona. Dos nuevos preparados de testosterona en gel han salido hace poco al mercado: Testogel® y Testim®. Contienen 1% de testosterona y liberan 50 mg de la hormona21. Antes de la aparición del gel se utilizaba la testosterona en parche escrotal (Testoderm®) o no escrotal (Androderm®, Testoderm TTS®). El parche escrotal es un copolímero de acetato de etilenvinilo que aplicado en el escroto libera de 4 a 6 mg de testosterona cada 24 horas. Por la vascularidad escrotal se obtiene una absorción excelente con niveles picos de testosterona en 2 a 4 horas. El testículo debe estar limpio, seco y afeitado. Uno de los problemas de este parche es la aparición de niveles muy altos de dihidrotestosterona por las concentraciones altas de la enzima 5 alfa reductasa en la piel del escroto, que convierte la testosterona en dihidrotestosterona. El parche de Androderm se aplica por la noche en el abdomen, muslos o brazos y libera 2.5 a 5 mg de testosterona. Produce más irritación de la piel que el parche escrotal y las concentraciones plasmáticas de testosterona son comparables. El Testoderm TTS® produce efectos similares al Testoderm® con menos irritación de la piel22. La testosterona se puede implantar en la piel en forma de semilla o cápsula. Con una dosis de 200 mg se puede mantener una concentración fisiológica de testosterona por 4 a 6 meses. Este método no ha recibido aceptación por requerir un procedimiento quirúrgico. La vía oral (metiltestosterona) no es muy utilizada por su poca efectividad y por su hepatotoxicidad. De reciente aparación son las tabletas mucoadhesivas de liberación sostenida que se absorben en la mucosa oral (Striant SR®), 30 mg diarios23. PRESENTACIÓN DEL CASO Paciente de 18 años que consultó en mayo de 2005 por falta de desarrollo sexual. Desde los 13 años notó genitales externos pequeños, ausencia de vello axilar y pubiano, disminución de la libido y disfunción eréctil. Presenta anosmia desde la infancia. Antecedentes y revisión de sistemas negativos. Al examen físico presentaba una talla de 163 cm y un peso de 51 kg. El resto de los signos vitales eran normales. Se notó la presencia de hábito eunucoide, atrofia testicular (volumen de 2 ml), pene pequeño (3 cm) y ausencia de barba, vello axilar y pubiano. No podía oler. El laboratorio mostró una testosterona sérica de 22 ng/dl (normal 241-827), FSH de 1 mUI/ml (normal 1.4-18.1), LH de 0.029 mUI/ml (normal 1.5-9.3), prolactina de 6.7 ng/ml (normal 2.1-17.7), tiroxina libre de 1.6 ng/dl (normal 0.89-1.76), hormona tiroestimulante (TSH) de 1.6 UI/ml (normal 0.35-5.5), 8 am cortisol de 10.3 mg/dl (normal 5-22), IGF-1 de 409 ng/ml (normal 239-630). Edad ósea de 14.6 años.
La historia, el examen físico y los datos de laboratorio, incluyendo la respuesta subnormal de FSH y LH a GnRH, son muy característicos del síndrome de Kallmann. Al paciente se le trató con 50 mg de enantato de testosterona cada 15 días con una muy buena respuesta en el desarrollo sexual, libido y erección. Hace poco el manejo se cambió a Androgel®). REFERENCIAS
© Copyright 2006 - Revista Colombia Médica |
| |||||||||