 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Revista Colombia Médica, Vol. 39, No. 3, Suppl. 3, Jul-Sept, 2008, pp. 38-45 El 17β-estradiol disminuye la expresión y asociación de quinasas responsables de la hiperfosforilación de Tau* β-estradiol decreases the expression and association of kinases responsible of Tau hyperphosphorylation Efraín Cepeda-Prado, MSc1, Alejandro López-Tobón, BSc2, Luis Miguel García-Segura PhD3, Gloria Patricia Cardona- Gómez, PhD4 * Investigación financiada por COLCIENCIAS código
11150416372 y la Universidad de Antioquia, Medellín, Colombia. Recibido para publicación diciembre 20,
2007 Code Number: rc08066 RESUMEN
Introducción: Un
componente molecular predominante en el estudio de las enfermedades
neurodegenerativas es la presencia del complejo Tau-GSK3β y su
asociación con agregados proteicos al interior de la
célula. Evidencias considerables muestran que GSK3β es el
principal causante de la hiperfosforilación de Tau. Sin embargo,
son poco claros los eventos moleculares que gobiernan este complejo. Palabras clave: 17 β-estradiol; Tau; β-amiloide; Neuroprotección; Plasticidad neuronal. LISTA ABREVIATURAS 17β-E2: 17β-estradiol. Aβ: Proteína β-amiloide. AMPA (ácido β-amino-3-hidroxi-5-metil-4-isoxazol-propiónico). Ca2+: Calcio. cAMP: Adenosin monofosfato cíclico. EA: Enfermedad de Alzheimer. FAK: Quinasa de adhesión focal. Fyn: Oncogen relacionado con SRC. GluR2/3: Subunidades 2/3 del receptor ionotrópico AMPA. GSK-3: Quinasa glucógeno sintetasa-3. LDH: Lactato deshidrogenasa. LY294002: Inhibidor específico para PI3K. MAPs: Proteínas asociadas con microtúbulos. NTF: en inglés Neurofiblillary tangle-Ovillos neurofibrilares. PHF: Filamentos helicoidales pareados. PP2: 4-amino5-(4-clorofenil)-7(t-butil)pirazol(3,4-D)piramidal. SH: Dominios homólogos a src. SRC: Gen con alta similitud con el sarcoma viral de Rous que codifica para una proteína tirosina quinasa. Tau: Proteína asociada con microtúbulos. SUMMARY
Introduction: A predominant molecular component analyzed in the study of neurodegenerative diseases is the presence of the Tau-GSK3β complex and its association with protein aggregation into the cell. Several evidences show that GSK3β
has an important role in abnormal pattern of the phosphorylation of
Tau. However, the molecular events that are governing this complex are
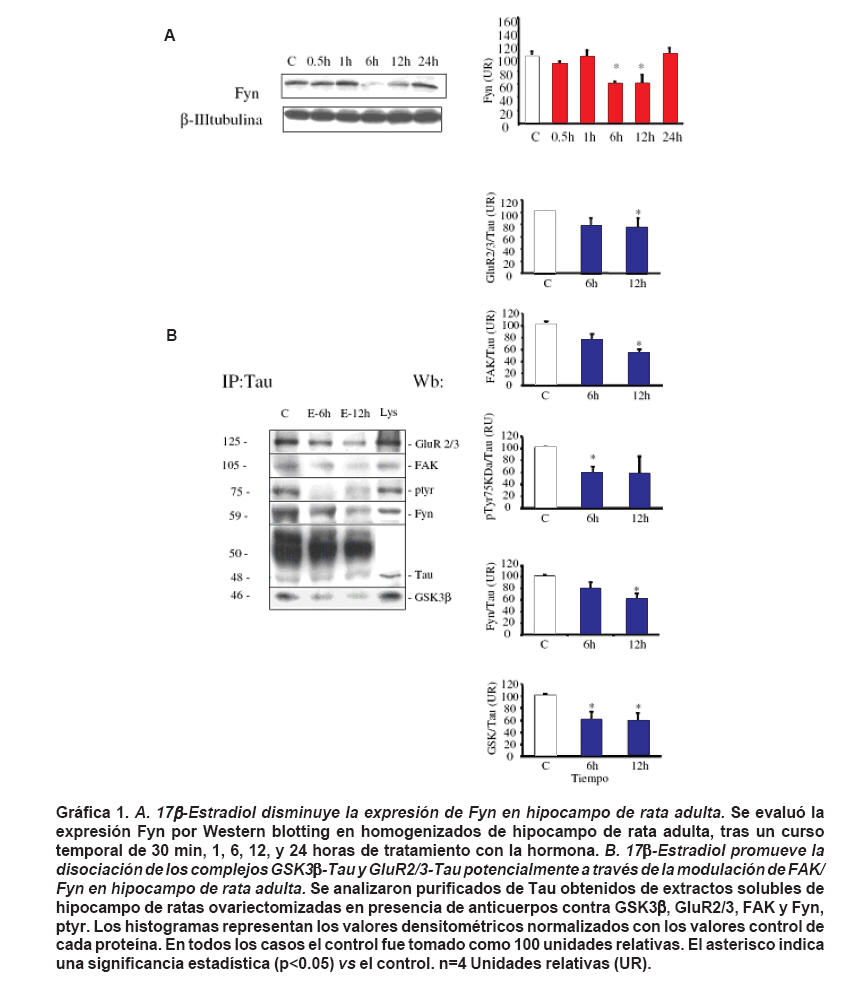
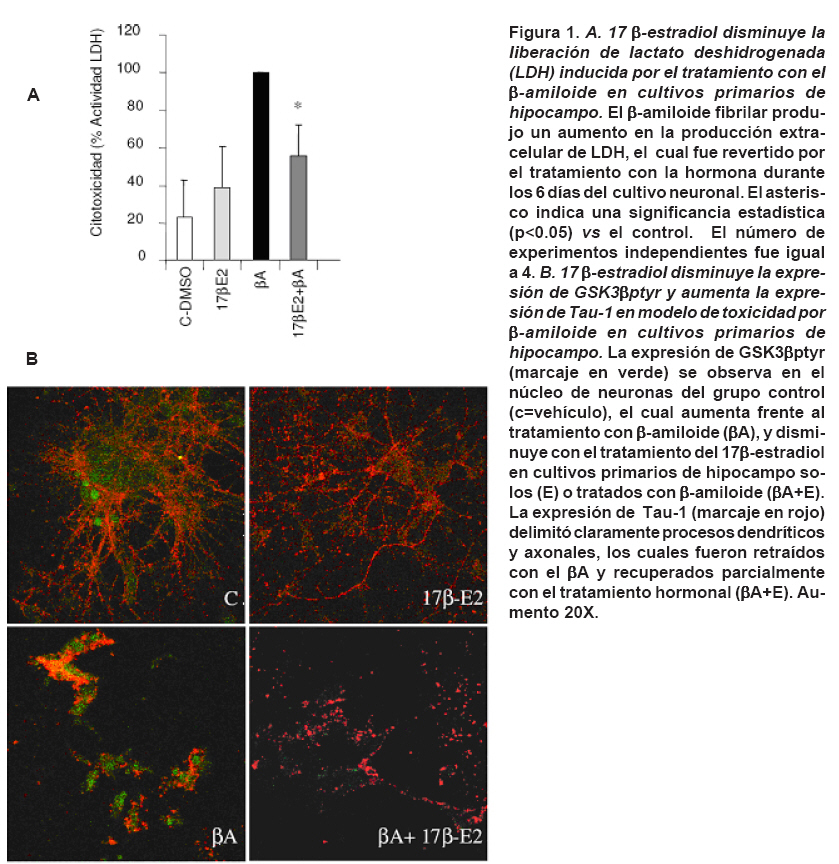
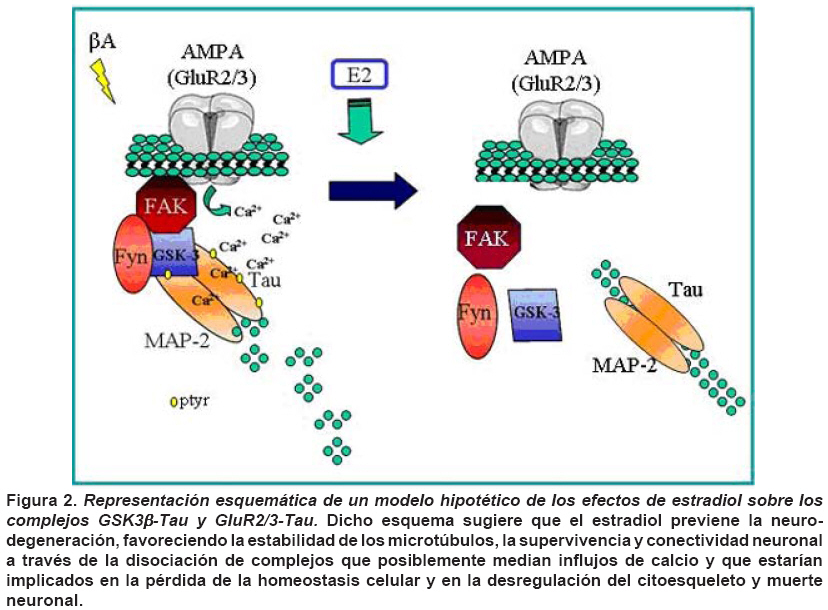
unknown. Keywords: 17 β-estradiol; Tau; β-amyloid; Neuroprotection; Neuronal plasticity. En las últimas décadas el estradiol ha adquirido una importancia crucial gracias a que puede interactuar con vías de señalización interviniendo con segundos mensajeros, entre estos Ca2+ intracelular y reguladores transcripcionales como el receptor de estrógenos1 y el cAMP2 . Además, promueve supervivencia neuronal, crecimiento y plasticidad neurítica tanto en modelos in vivo como in vitro, convirtiéndose en un blanco para el desarrollo de estrategias farmacológicas que estimulen la supervivencia neuronal y reconectividad sináptica1,3-5. Las enfermedades neurodegenerativas se caracterizan por ambientes citotóxicos que generan una pérdida neuronal selectiva atribuida a la presencia de inclusiones citoplasmáticas. Particularmente, la enfermedad de Alzheimer (EA) se caracteriza por la formación de placas seniles, constituidas por beta-amiloide (βA) y conglomerados neurofibrilares (neurofiblillary tangle, NFT), propiciados por Tau hiperfosforilada formando estructuras aberrantes filamentosas conocidas como filamentos helicoidales pareados (PHF; paired helical filaments)6,7. La quinasa glucógeno sintetasa-3 (GSK3) es una proteína serina/treonina quinasa que está implicada en múltiples procesos celulares, su inhibición permite la síntesis del glicógeno, proteínas y activación de factores de transcripción implicados en supervivencia celular8. Hoy en día, GSK3 fosforilado en tirosina (ptyr) representa uno de los principales causantes de la fosforilación anormal de Tau9. Sin embargo, hasta ahora no se han esclarecido los componentes moleculares que participan en la formación del complejo GSK3-Tau. Estudios previos4,10 indican la existencia de este complejo que potencialmente podría ser removido en presencia de estradiol favoreciendo la remodelación del citoesqueleto en procesos de neuroprotección4,11. Además, se ha visto que la conformación GSK3-Tau esta íntimamente relacionada con proteínas asociadas con microtúbulos y estos acoples al parecer estarían regulados por la pre-sencia de quinasas. Entre ellas se encuentra FAK y Fyn proteínas comprometidas en la adhesión celular y remodelamiento del citoesqueleto neuronal12. Por tanto, en el presente estudio se evalúo el efecto del 17 β-estradiol sobre la expresión de Fyn y la inter-acción de Tau con ésta y otras proteínas como GSK3β-ptyr relacionadas con la hiperfosforilación de Tau y su potencial correlación en un modelo de toxicidad por β-amiloide. MATERIALES Y MÉTODOS Animales y tratamiento hormonal. Se utilizaron ratas hembras albinas de la cepa Wistar del Bioterio SPF de la Sede de Investigación Universitaria de la Universidad de Antioquia, llevando a cabo las normas de mantenimiento en el bioterio bajo condiciones controladas de temperatura (22ºC), humedad y fotoperíodo luz/oscuridad de 12:12 h y recibieron alimentación y agua ad libitum. Los animales se manipularon según las consideraciones éticas dispuestas en la Resolución N° 008430 de 1993 del Ministerio de Salud y en la Ley 84 de 1989, teniendo en cuenta una orientación bioética, con un cuidado especial, para minimizar el sufrimiento de los animales y reducir el número de ratas usadas. Las ratas previamente se ovariectomizaron al mes de edad, anestesiadas intraperitonealmente (ip) con 2,2,2, tribromoetanol al 2% (0.2 g/kg bw, Fluka Chemika AG, Buchs, Swizerland). Un mes después se trataron con 17 β-estradiol (300 mg/rata, Sigma) vía intraperitoneal13. Luego los animales se sacrificaron a las 6 y 12 h, y los cerebros se extrajeron para el análisis bioquímico. Cultivos primarios. Las neuronas hipocampales de embriones de ratones E18 provenientes de ratas Wistar se cultivaron en medio Neurobasal-B27 (GIBCO), suplementado con 25 mM KCl y 1 mM glutamina. Estas células fueron plaqueadas dentro de platos de 24 pozos recubiertos con poly-D-lysina/laminina a una densidad de 2-6 x 105 células/pocillo. Las células fueron lisadas a los 6 días después del tratamiento hormonal para su posterior análisis. Para cada grupo experimental in vitro se usó un n de 3, realizando triplicados. Western blotting. Los animales se sacrificaron y sus cerebros se removieron rápidamente. Los hipocampos se disecaron y homogenizaron en tampón de lisis que contenía 150 mM NaCl, 20 mM Tris, pH 7.4, 10% glicerol, 1 mM EDTA, 1% NP40, 100 mM fenilmetilsulfonil fluoro, 1 mg/ml aprotinina y leupeptina (Sigma) y 100 mM vanadato. Se llevaron a cabo electroforesis en geles de poliacrilamida (10%-12% SDS-PAGE) usando un sistema mini-protein (Bio-Rad); se cargaron 30 mg de proteína en cada uno de los carriles acompañados de buffer de carga que contenía 0.375 M TRIS, pH 6.8, 50% glicerol, 10% SDS, 0.5 M DTT y 0.002% de azul de bromofenol. Las muestras se calentaron a 100ºC por 3 min y luego se cargaron en el gel. Después de la electroforesis, las proteínas se transferieron a membranas de nitrocelulosa (Amersham) a través de un sistema electroforético (mini Trans-blot Electrophoretic Transfer Cell) a 250 mA por 2 h. Las membranas se lavaron tres veces por 5 min con TTBS (Tris-HCl 20 mM pH 7.5; NaCl 500 mM; 0.05% Tween-20 que contenía solución salina TRIS-buffer, pH 7.4), incubadas con leche descremada en polvo al 5% por 30 min y finalmente se realizaron varios lavados con TTBS e incubadas toda la noche con los diferentes anticuerpos a una temperatura de 4Cº. Se usaron los siguientes anticuerpos: anticuerpo monoclonal hecho en ratón para GSK-3β (Transduction Laboratories, diluido 1:2500), Tau, Fyn, FAK (Santa Cruz Biotechnology 1:1000) y GluR2/34 . Los lisados totales se usaron como control positivo. Al día siguiente las membranas se lavaron e incubaron con los anticuerpos secundarios (anti-ratón IgG o anti-conejo IgG, conjugados con peroxidasa; Jackson InmunoResearch Laboratories, diluídos 1:20,000) por una hora a temperatura ambiente. La señal inmunorreactiva se obtuvo usando quimioluminiscencia (ECL Western Blotting System, Amersham) seguido por una exposición a películas de autorradiografía (Hyperfilm ECL, Amersham), las cuales se analizaron usando un programa de cuantificación (QUANTITY ONE Quantification Software, versión 4.3.0.) y el uso de un calibrador densitométrico (BioRad, GS800 Calibrated Densitometer). Los resultados de cada una de las membranas fueron normalizados con los valores del control (ratas ovariectomizadas tratadas con vehículo). Todas las muestras se procesaron en paralelo en busca de minimizar las variaciones entre los ensayos. Co-inmunoprecipitación. Los lisados se centrifugaron a 14,000 rpm por 2 min, 1 mg de extracto se incubó toda la noche a 4ºC en presencia de un antisuero policlonal contra Tau (Santa Cruz Biotechnology, diluido 1:250). Se adicionaron perlas de proteína A sefarosa y se incubaron durante 2 h. Los inmuno-complejos se lavaron tres veces con buffer de lisis de inmunoprecipitación y después se analizaron por Western blot. Ensayo de toxicidad. Para cuantificar la muerte celular se midió la liberación de lactato deshidrogenasa (LDH) a los 6 días de tratamiento con el β-amiloide y el tratamiento con el 17 β-estradiol en cultivos primarios de hipocampo. Se realizó un ensayo colorimétrico con relación a la cantidad de sales de formazan generadas después de la conversión del lactato a piruvato, por reducción de sales de tetrazolium, la cual es proporcional a la actividad de LDH en la muestra. Luego los sobrenadantes libres de células se colectaron y se incubaron a temperatura ambiente durante 60 min con una mezcla de reactivo apropiado de acuerdo con las instrucciones del estuche de excitotoxicidad de Roche® (Cytotoxicity Detection Kit, Roche, Switzerland). Así, de acuerdo con la intensidad del color rojo formado y la longitud de onda de 490 nm en la que se leyeron las placas, la valoración fue proporcional a la actividad de LDH y la cantidad de células dañadas. Los datos se normalizaron con la actividad de LDH del tratamiento con β-amiloide, el cual correspondió al 100% y se expresaron como un porcentaje con respecto a este tratamiento ±SEM establecido de un n de tres experimentos independientes. Microscopía confocal. Los cultivos primarios se fijaron en parafolmaldehído al 4% durante 20 min y luego lavados y preincubados con PBS conteniendo 0.1% de Triton X-100 y 1% de suero bovino (PBS-BS). Luego, se incubaron con anticuerpos primarios diluidos en PBS (anti-GSK3btyr216, anti-Tau-1), por 2 h a RT o toda la noche a 4ºC. Después de lavados seriados las células se incubaron con anticuerpos secundarios conjugados con fluoróforos Alexa 488 y Alexas 594 (Molecular Probes). Las células se lavaron cuatro veces en tampón y se cubrieron con solución de montaje gerbatol, y se observaron por microscopía confocal en el Instituto Cajal, del CSIC, Madrid España. Las imágenes de confocal se analizaron como imagen individual, colocalizadas e integradas en tres dimensiones, para analizar la localización subcelular de las proteínas diana. Se realizaron controles omitiendo incubación con anticuerpos primarios para evaluar especificidad. Análisis estadístico. Cada grupo experimental contó con un «n» de 3-5. Debido a que la prueba Fischer demostró una varianza significativamente homogénea entre los grupos, se seleccionó la prueba de análisis de varianza de una sola vía (ANOVA) para estadística de comparaciones múltiples. La prueba de Tukey post-hoc se usó para determinar diferencias significativas entre dos grupos independientes. Se adoptó nivel de confianza de p<0.05. Los datos fueron expresados como el promedio más el error estándar medio (± SEM en sus siglas en inglés). RESULTADOS 17 β-estradiol modula la expresión de Fyn y su interacción con Tau. La expresión de Fyn disminuyó en el hipocampo de ratas tratadas con 17 β-estradiol a las 6 y 12 horas tratamiento (Gráfica 1A). Adicionalmente, el tratamiento hormonal disminuyó la asociación entre FAK y Fyn con Tau a las 12 horas, acompañado de la disminución de fosforilación en tirosina de Tau (Gráfica 1B). 17 β-estradiol disminuyó la conformación del complejo GSK3β-Tau y GluR2/3-Tau. Inmunoprecipitados de Tau obtenidos de muestras de hipocampo de ratas ovarectomizadas tratadas con 17 β-estradiol, mostraron una disminución en los niveles de Tau-GSK3β y Tau-GluR2/3, al analizar los datos obtenidos durante intervalos de 6 y 12 horas de tratamiento (Gráfica 1B). 17 β-estradiol protege contra la toxicidad del β-amiloide. En cultivos primarios de hipocampo de rata, el estrógeno disminuyó significativamente los niveles de lactato deshidrogenasa liberados al medio por las neuronas tras el tratamiento con el β-amiloide. El tratamiento con el β-amiloide produjo niveles altos considerados el 100% de expresión respecto al control negativo y al tratamiento con 17 β-estradiol (Figura 1A). Además, las neuronas tratadas con β-amiloide, la expresión de GSK3β-ptyr aumentó y la morfología de Tau presentó un aspecto de acumulación y desarreglo del citoesqueleto; sin embargo, el tratamiento con estrógeno disminuyó la expresión de GSK3β-ptyr, favoreció la expresión de Tau defosforilado a lo largo del axón en el tratamiento solo con la hormona y frente a la toxicidad del β-amiloide, aunque este último fue menos evidente (Figura 1B). DISCUSIÓN Los resultados del presente artículo muestran aspectos moleculares nuevos que sugieren un efecto modulador del 17 β-estradiol sobre la formación de complejos Tau-GSK3β y Tau-GluR2/3, potencialmente regulados a través de Fyn. Adicionalmente, estos resultados confirman una vez más cómo el efecto neuroprotectivo de la hormona podría ser mediada a través de la regulación del estado de fosforilación de la GSK3β. Como se mencionó antes, una de las características de la participación de Tau en enfermedades neurodegenerativas es la desregulación en la fosforilación de la proteína14. El ensamblaje y desensamblaje de microtúbulos es regulado por diferentes patrones de fosforilación de Tau, que resultan en una mayor o menor afinidad por estos9. También se ha observado un incremento en la fosforilación de Tau y MAP2c, junto con la presencia PHF, en cultivos primarios de neuronas corticales de seres humanos y ratas tratados con βA. Estos cambios fueron atenuados por la adición de 4-amino5-(4-clorofenil)-7(t-butil)pirazol(3,4-D)piramida (PP2) un inhibidor de tirosin quinasas de la familia Src e igualmente sucedió con LY294002, un inhibidor de PI3K. Además, en este mismo estudio observaron que el aumento en la fosfoliración de Tau y MAP2c se correlaciona con un aumento en la actividad de la quinasa de adhesión focal FAK y Fyn15. Adicionalmente, algunos estudios7,12 señalan una interacción de tirosina quinasa de la familia src con Tau, sobre todo por la presencia del sito de PXXP que interactúa directo con los dominios SH2 y SH3 que es bastante conservado en esta familia. Un ejemplo de esto, es la relación de Fyn con Tau en los eventos de mielinización llevados a cabo por oligodendrocitos durante el neurodesarrollo16. Un hecho, que hace aún más interesante las interacciones entre Tau y quinasas citoplasmáticas es la respuesta de hiperfosforilación de Tau en cultivos tratados con Ab, donde se demuestra que esta hiperfosforilación se debe a la presencia de asociaciones entre FAK/Fyn que eleva la fosforilación en tirosina afectando sobre todo proteínas asociadas con microtúbulos15. Este último estudio apoya los resultados de este artículo, porque se ha visto una disminución de los niveles de Fyn a las 6 y 12 h (Gráfica 1A) que se correlacionan con la disociación de Tau y GSK3β en estos mismos tiempos tras el efecto del 17 β-estradiol, acompañados de una disociación de Tau con FAK y Fyn y una disminución en el estado de fosforilación Tau en tirosina (Gráfica 1B), lo que sugiere que esas quinasas podrían ser reguladas tras eventos de plasticidad neuronal y remodelamiento sináptico ejercido por la hormona en condiciones basales. Por otra parte, es clave resaltar la importancia de los presentes hallazgos con respecto a las relaciones existentes entre el complejo Tau-GSK3β y Tau-GluR2/3, y alteraciones en la concentración del calcio intracelular y la despolimerización de microtúbulos en eventos de retracción o más severamente frente a eventos de excitotoxicidad como el producido por el β-amiloide. En este sentido se ha observado que la desregulación de la expresión de GluR2 produce un aumento en el influjo de Ca2+ a través de receptores alpha-amino-3-hidroxi-5-metil-4-isoaxazolepropionato (AMPA) que son activados por glutamato, por lo general implicados en desregulación por isquemia cerebral que se caracteriza por una disminución en la población de neuronas piramidales en las regiones CA1 y CA3 del hipocampo17 y que en este modelo podrían ser desrregulados por la toxicidad del β-amiloide (Figura 1A). Recientemente se ha observado que dicha toxicidad disminuye la expresión de GluR2 y calcineurina, asociados con estrés oxidativo y cambios en la concentración intracelular de Ca2+18,19. Dicho influjo de calcio genera una distorsión en el transporte axonal propiciado por la alteración de la polimerización de microtúbulos y presencia de agregados20, lo cual parcialmente podría explicar el aspecto de acúmulos en la conformación de Tau después del tratamiento con el péptido amiloide fibrilar (Figura 1B), así como la retracción de dendritas y axones observados en las neuronas hipocampales, los cuales fueron parcialmente revertidos por el tratamiento con el 17 β-estradiol (Figura 1B), cuyo tratamiento también previno la toxicidad al disminuir la liberación de la lactato deshidrogenada (Figura 1A). Basados en lo anterior, es posible sugerir que la formación del complejo Tau-GSK3β y -Tau-GluR2/3 se puede encontrar potencialmente vinculado a un aumento en el influjo de Ca2+ que de modo transitorio se daría en la retracción de dendritas y axones en eventos de remodelamiento del citoesqueleto neuronal y de modo más severo y permanente estaría asociado con eventos de estrés y muerte celular por estímulos excitotóxicos. Sin embargo, el estradiol es capaz de generar una disociación de esas interacciones macromoleculares posiblemente reestableciendo la homeostasis intracelular con ayuda de quinasa reguladoras como GSK3 y Fyn, al disminuir la expresión de GSK3βptyr y al aumentar el estado de Tau defosforilado (Tau1) en cultivos primarios de hipocampo (Figura 1 y 2). Estos resultados son apoyados por estudios previos en los que se ha observado un efecto neuroprotectivo del estrógeno frente a isquemia cerebral tras la disminución de la hiperfosforilación de Tau (PHF) y el aumento de GSK3β fosforilada en serina 9, la cual está involucrada en supervivencia celular11. Adicionalmente, estos efectos podrían estar apoyados con el efecto del estrógeno en condiciones basales tras 12 horas de tratamiento en hipocampo de rata adulta, en el cual produjo la disociación completa de los complejos de Tau-Fyn y Tau-FAK. En resumen, estos hallazgos sugieren que el 17 β-estradiol ejerce su acción neuroprotectiva a través de la disminución de la formación de complejos moleculares asociados con citotoxicidad y al disminuir la expresión y asociación de quinasas responsables de la hiperfosforilación de Tau, que a su vez reestablecerían la polimerización de microtúbulos a través de la disminución del influjo de Ca2+, evitando la formación de inclusiones citoplasmáticas clásicas de los procesos de neurodegeneración. Sin embargo, es necesario estudios complementarios que permitan ahondar en los mecanismos de regulación y proponer potenciales nuevas dianas terapéuticas. REFERENCIAS
© Copyright 2008 - Revista Colombia Médica The following images related to this document are available:Photo images[rc08066f1.jpg] [rc08066f2.jpg] [rc08066g1.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}