 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Revista Colombia Médica, Vol. 41, No. 1, 2010, pp. 76-81 Glycogen storage disease: report of two cases in the city of Cartagena Enfermedades de depósito de glucógeno: informe de dos casos en la ciudad de Cartagena Ciro C. Alvear, MSc1, Miriam Barboza, MD2, Zeudy K. Rodríguez, MD3
1Clinical Biochemistry, Professor Faculty of Medicine, Universidad
de Cartagena, Cartagena, Colombia.
e-mail: cicealse@yahoo.es Received
for publication December 1, 2008 SUMMARY Objective: to
report two cases of children with type Ia glycogen storage disease compatible
with Von Gierke disease, suspected in the presence of findings such as hepatomegaly,
nephromegaly, hypoglycemia, and stunted
growth. Keywords: Hepatomegaly; Nephromegaly; Hypoglycemia; Stunted growth; Metabolic screening; Glucose-6-Phosphatase; Diet. RESUMEN Objetivo: Comunicar
dos casos de niños con glucogenosis compatibles con el tipo Ia o enfermedad
de Von Gierke, que se debe sospechar ante la presencia de hallazgos como hepatomegalia,
nefromegalia, hipoglicemia y
talla baja. Palabras clave: Hepatomegalia; Nefromegalia; Hipoglicemia; Talla baja; Retardo en el crecimiento; Tamizaje metabólico; Glucosa 6 fosfatasa; Dieta. Glycogen storage disease (GSD) or glycogenosis include hereditary diseases caused by abnormalities of the enzymes that regulate the synthesis and degradation of glycogen. Glycogen synthesis is produced in numerous tissues, especially in the liver, kidneys, and muscle; and mainly deposited in the liver and muscle, but the latter lacks glucose-6-phosphatase, an enzyme that acts in the light of the endoplasmic reticulum and turns glucose-6-phosphatase into free glucose2,3 and, hence, insufficient amounts of glucose is liberated onto the systemic circulation given that the function of muscular glycogen is to serve as a source of energy for its own activity. The clinical manifestations of diseases with glycogen storage are often due to hypoglycemia with or without increased glycogen storage. The location of the enzymatic blockage along the metabolic path determines if the glycogen configuration is normal or abnormal2,3. In general, a numbering system from 0 to XII has been accepted for these disorders; additionally, these can also be classified according to the affected organ and the clinical manifestations in hepatic and muscular glycogenosis1-4. The object of this presentation is to report the cases of two siblings with different clinical and biochemical manifestations of glycogenosis, which because of its characteristics corresponds to the type I or Von Gierke’s disease. Photographs were taken after securing a signed informed consent, as registered in the clinical histories of both child patients, to document that exposed by the physical exam. FIRST CASE The case of an 11-year-old male patient,
attending school from the city of Cartagena, Colombia. The history of his illness
began at 11 months of age when the mother consulted because the child presented
a crisis with generalized cyanosis without other aggregate symptomatology,
discarding cardiac pathology. As important antecedents, the child is
the product of a second
pregnancy, with good apgar at birth. He was breast fed until two years of age.
He revealed normal language and psychomotor development. As important personal
antecedents, according to the mother an older brother also presented the same
symptoms. Three months after the initial consultation, the patient presented
generalized tonic-clonic crisis with a normal physical examination. At two
years of age, the mother again consults because the child presented abdominal
distension,
weight loss, asthenia, adynamia, and generalized paleness; followed by diarrhea
and abdominal pain. The physical exam at the time found generalized paleness,
globe-like abdomen due to 4-cm hepatomegaly and ascytis. The following tests
were done: hemogram, platelets, TP, TPT, proteinogram, and glycemia, normal.
Serology for hepatitis B was negative. Metabolic screening was initially normal;
although, a second exam revealed positive Benedict, Seliwanoff, and nitrosonaphthol
tests. Based on this, it was felt that the child could have congenital galactosemia
and a diet was indicated for this entity. At 4 years of age, the child again
presented a generalized tonic-clonic crisis associated with vomiting and the
clinical exam revealed a 5-cm homogenous hepatomegaly. Hypoglycemia with a
level of 31 mg/dl was detected. A cerebral CT scan was ordered and it
was reported
normal. A carpogram was carried out at that moment corresponding to an osseous
age of 4 years and 9 months. Further tests: TSH, T4 L were within normal limits.
AST, alkaline phosphatase and LDH elevated at 829 U/l (Normal value: 160-320
U/l). The physical exam found the
patient’s weight at 15 kg (p10), height 88 cm ( At 5 years of age, the
patient presented persistent hematuria that remained with periods of asymptomatic
intermittent hypoglycemia. At this age, nephromegaly was detected and a hepatic
biopsy was practiced, which reported hepatic tissue with marked hepatocyte
distension; with special tinctures it was possible to identify much of the
positive PAS glycogen
of normal structure, which is completely digested with the Diastase PAS. There
was no change in fatty tissue. Electronic microscopy found displacement of
organelles toward the periphery and a focal presence of glycogen granules in
cytoplasm;
these changes are compatible with type I glycogenosis. At 9 years of age, persistent
hepatomegaly and nephromegaly were found; additionally, he fractured his left
arm and it
did not consolidate well. The patient is currently
11 years old, 120 cm tall ( SECOND
CASE 14-year-old male patient, studying in
the 9th grade from the city of Cartagena, Colombia. He was remitted at 8 years
of age to the Biochemistry unit due to family history of glycogen storage disease
(first case). His illness began at
18 months of age with abdominal distension, aqueous diarrhea with a month and
a half evolution, generalized weakness, and progressive edema on face and feet,
and paleness of the skin and mucous membranes. Important antecedents: product
of the first pregnancy of non-consanguineous parents, mother G2 P2 A0 C1, normal
vaginal birth, pregnancy free of complications, and normal apgar at birth.
His psychomotor development was normal; was breast fed by his mother until
he was
two years old. There is no history of epileptic crises. The physical exam found:
diffuse hepatomegaly without pain 2 cm below the right costal and anasarca
(extreme generalized edema) flange. A neurological exam proved normal. At this
age, the
patient was subjected to hepatic tests, as well as lab exams for cholesterol,
triglycerides, and uric acid; all were found within the upper normal limit.
No data on hypoglycemia are presented, the Beneditt and Seliwanoff tests were
positive,
while tests for mucopolysaccharidosis resulted negative. Gammography and hepatic
ultrasound tests revealed diffuse hepatomegaly. Renal ultrasound did not show
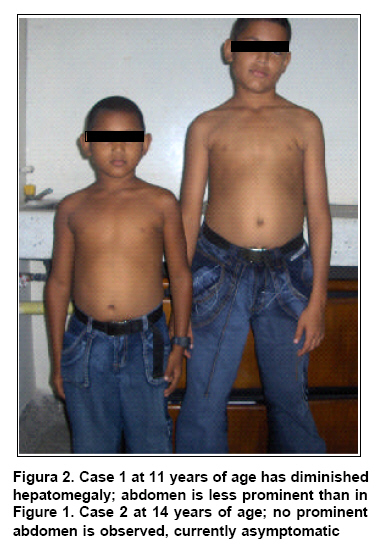
alterations. Symptoms remitted at 5 years
of age (Figure 2). The physical exam carried
out upon consultation when he was 8 years old was normal, and currently there
have been no modifications of such. DISCUSSION Herein, we present two cases of glycogenosis
in siblings with different biochemical and clinical expressions, is spite of
their consanguinity. The renal and hepatic compromise and the demonstration
of hypoglycemia in the younger brother (first case), which led him to presenting
an epileptic crisis of generalized tonic-clonic type makes us consider that
the
type of glycogenosis is Von Gierke type I added to the fact that in the hepatic
biopsy the glycogen storage was detected but with normal structure. The second
case did not document symptomatic hypoglycemia or alterations in
lactic acid. The other glycogen storage
diseases that present a normal glycogen structure are1-4: Statistically, glycogenosis has a global prevalence of 1:20.000 to 1: 25.000
live births, with types I a, I b, II, III, and VI being the most common with
90% of the cases2-4. Glycogenosis type I or
Von Gierke’s disease, was discovered by German physician Gierke, who studied
an 8-year-old girl with chronic increase
in liver size. After the girl’s death in 1929, due to a common cold, it
was proven that her liver contained 40% glycogen. The glycogen appeared normal,
but it could not be degraded by the girl’s liver extracts, but it could
be degraded by extracts from other livers2,3. It is the most common
hereditary diseases of type I glycogen storage and, clinically, the most serious,
where the cause of the disturbance is the absence of or diminishing of glucose-6-phosphatase
of the liver, the
intestinal mucosa, and the kidneys2,3; leading to an accumulation
of abnormally high amounts of glycogen in tissues like the liver and renal
tubular cells, hence, through this mechanism producing
hepatomegaly and nephromegaly3,4, the first is well-described in
both patients previously discussed and nephromegaly was noted in the younger
patient. Type I glycogenosis is
inherited with recessive autosomal character. The gene for glucose-6-phosphatase
is located in chromosome 17q21. The most frequent mutations responsible for
this illness are known and it is possible to detect carriers and conduct prenatal
diagnosis through DNA diagnostic tests. The genetic anomaly in the hydrolysis
of
glucose-6-phosphatase only appears once per every 200,000 individuals4,5. Patients with type I
glycogenosis may be classified into various subtypes, with the most common
belonging to those who lack the glucose 6-phosphatase
enzyme per se, or Von Gierke’s disease (Type I a)5. Here,
glycogen is synthesized normally and, thus, its structure is normal; but there
is a failure
in glycogenolysis which impedes the liberation of glucose from these deposits.
In the less serious expression of this disease, the concentrations of blood
glucose are normal except after stressful situations, where the normal hyperglycemic
response is inhibited2,3. The liver in these patients
liberates some glucose, through the action of the debranching enzyme that helps
to complete the process of glycogen hydrolysis by allowing the glycogen phosphorylase
to continue degrading
glycogen2. Overproduction of purine
and hyperuricemia in Von Gierke’s disease are secondary effects to the
greater generation of the PRPP precursor, ribose-5-phosphate. A secondary lactic
acidosis raises the renal
threshold for urate, which leads to hyperuricemia2-4. This stems
also form the increased degradation of purines in the liver; hyperlipidemia
is due
to the increase in the availability of lactic acid for lipogenesis and to the
mobility of lipids from the adipose tissue, provoked by elevated levels of
glucagon, which are a response of hypoglycemia. Hyperuricemia occurs in small
children,
but gout is
usually not manifested prior to puberty2,3. During physical exam,
patients tend to have fat cheeks, thin extremities, short height, and protuberant
abdomen due to massive hepatomegaly3,4. Although the phenotype of
the patients does not correspond to what has been described, it can be noted
that both are short for their ages, and with the first case revealing affectation
to osseous age. The kidneys also show increase in size, as with the child in
the case where nephromegaly was detected via ultrasound at 5 years of age;
while the
spleen and the heart are normal. Pulmonary arterial hypertension
has been described in type Ia, possibly due to an abnormally excessive production
of vasoconstrictor amines like serotonin that is synthesized from the intestinal
enterochromaffin cells. Hence, this becomes a plausible means in studying patients
suspected of having type Ia glycogenosis; however, we should not discard the
effect caused by the endothelial damage from the presence of metabolic alterations
like hyperlipemia3,4. The clinical manifestations
are given by hypoglycemia in fasting state that can lead to epileptic crisis,
which are frequent and almost invariably represent the initial disorder in
children affected, as presented by the child in the first case who in two opportunities
manifested generalized tonic clonic crisis without an apparently unchaining
factor
and with a normal physical exam. Hypoglycemia can also evolve to chronicity,
as a consequence of insufficient enzyme required to obtain glucose from hepatic
glycogen and from gluconeogenesis; this also occurred with the first child.
Although type I glycogenosis mainly affects the liver, multiple organic systems
tend to
be involved. Puberty tends to be delayed in this disease. Frequent fractures
and radiological signs of osteopenia are not rare in adults and the mineral
content of radium is significantly reduced in pre-puberty patients; this would
be the
cause of the fracture noted in the first patient. Hematomas and epistaxis are
frequent and are associated to prolonged coagulation times consequential of
the alteration of platelet
aggregation and adherence2-4. Among the laboratory
findings, besides hypoglycemia, we can find: lactic acidosis, ketosis and hyperlipidemia,
this last one predisposes a greater risk for pancreatitis, atherosclerosis,
and cerebrovascular
events3,4. Lactic academia appears because the liver cannot effectively
use lactate to synthesize glucose. Additionally, in response to glucagon the
liver produces even more lactic acid. This hormone should unleash the liberation
of glucose without lactate production; nevertheless, the contrary occurs given
the lack of glucose-6-phosphatase. Anemia may be present and it is of multifactorial
etiology, contributing factors like recurrent chronic infection, intestinal
inflammatory disease, iron, vitamin B12, or folic acid nutritional deficiencies.
Furthermore,
stunted growth can be provoked by excessive counter-regulating hormones like
cortisol, which
is secreted during chronic hypoglycemia3-5. Patients with type I
glycogenosis may debut during the neonatal period with hypoglycemia and lactic
acidosis and even hepatomegaly; however, this disease generally tends to appear
at 3-4 months of age with
hepatomegaly or hypoglycemic crisis5. Although it is true that the
age of onset of the symptoms can be as early as the neonatal stage, the two
cases reported went without symptoms until the late lactating stage, thus,
the clinical
alterations appeared at that age. It must be indicated that these children
were breast fed up to two years, which could have favored the delay in the
onset of
the symptoms
because they received fractioned feedings. The gravity of the disease
reaches a plateau after the fourth or fifth year of life, as with the older
sibling who did not show symptoms until he was five years old. But other patients
present
by this age hepatic adenomas that may bleed and that in some cases turn into
malignant neoplasias. For this, currently makers are used like globular sedimentation
velocity (GSV) and alkaline phosphatase that frequently increase in the presence
of adenomas, as well as serum a-fetoprotein that is elevated exclusively in
cases of hepatocellular carcinoma1. Other complications are pulmonary
hypertension and nephrocalcinosis, nephrolithiasis and nephropathy that lead
to renal failure,
requiring
dialysis and renal transplant3,4. If we note in the first case,
in spite of transpiring with hepatic compromise for 9 years, tumors have not
been
documented at that level nor have there been complications of renal origin.
When the children with this disease mature, they become normoglycemic and characteristically
present abnormal glucose tolerance curves. This is why the carbohydrates must
be watched in the diet, given that excessive glucose leads to glycogen storage
in the liver and kidneys3,4. This characteristic may be observed
in the first case after starting treatment with cornstarch as of 4 years of
age
and with the passage of time, the diet is now normal and the 11 year-old boy
currently has not presented any
hypoglycemia crisis. The diagnosis of type
I glycogenosis is suspected because of the clinical presentation and the presence
of abnormal concentrations of lactate and lipids. Administering glucagon or
adrenalin determines a small or nil increase of the glycemia, while the concentration
of
lactate increases considerably. The definite diagnosis requires a hepatic biopsy
to demonstrate the deficit of enzymatic activity, although the enzymatic activity
can also be measured in the peripheral leucocytes and in a biopsy if the small
intestine. Identifying mutations of genes of glucose-6-phosphatase offers a
non-invasive diagnostic method for most
patients with type I glycogenosis3. In the cases described, it was
not possible to conduct a measurement of the enzymatic activity, although with
the patient of the first case an anatomopathological and histological study
was conducted with the corresponding tinctures, which allowed proving glycogen
storage
with normal structure. If we correlate clinical aspects with the cardinal symptoms
of hypoglycemia associated to hepatomegaly and nephromegaly, we should consider
a
glycogen storage disease: Von Gierke’s disease. The treatment is designed
to keep glycemia normal, which is accomplished through continuous infusion
of glycosides in the intestine through a nasogastric intubation or oral administering
of raw cornstarch; thus, alleviating the symptoms and avoiding hypoglycemia.
The treatment with cornstarch is based on the fact that the starch is hydrolyzed
by the intestinal glucoamylase and the pancreatic amylase and it is useful
in
older children, adolescents, and adults with a dosage of 1.5 to 2 g/kg/day,
which aids in keeping the level of glycemia normal for a period of 4 to 10
hours, as
long as the initial glycemia was normal. For a better effect in this therapy,
the cornstarch should be raw, mixed with cold water to keep the granules from
hydrolyzing and making the treatment less effective. In some patients, some
secondary effects may be noted like occasional diarrhea, abdominal distension,
and flatulence,
but these are usually transitory3-5. Because fructose and
galactose cannot directly become glucose, their ingestion should be restricted
and it is recommended to administer calcium and
multivitamin supplements3-5. Dietary treatment improves
hyperuricemia, hyperlipidemia, and renal function, preventing renal insufficiency.
However, after puberty the use of
allopurinol and hypolipemiants2 is required. Bearing in mind that
both children are currently in their adolescent stage, the levels of uric acid
and lipids have to be watched for early detection of any metabolic alteration
of this type. Microalbuminuria is treated with angiotensin converting enzyme
inhibitors (ACEI) like captopril. Citrate supplements are beneficial to prevent
or improve nephrocalcinosis and
the development of kidney stones2-4. As an alternate treatment,
hepatic or hepatocytes transplant and transposition of the portal vein have
been undertaken, creating a portacaval shunt that increases peripheral blood
glucose
by keeping portal blood from going into the liver. However, given complications
inherent to this treatment in the short and long term, it is recommended only
for patients with hepatic cancer3,4. As for the prognosis,
it is known that long-term complications tend to appear in adults whose illnesses
have not been treated adequately during childhood; currently, early diagnosis
and efficient treatment have notably improved the result. Nevertheless, nephropathy
and the
formation of hepatic adenomas continue being serious complications1,5. RECOMMENDATIONS REFERENCES © Copyright 2010 - Revista Colombia Médica |
| |||||||||
{kind=link}