 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Colombia Médica, Vol. 41, No. 4, October-December, 2010, pp. 388-395 The L-Arginine-Nitric Oxide-Peroxynitrite pathway (LANOP pathway): Does it protect or worsen the course of Chagas disease? Vía L-Arginina-Óxido Nítrico-Peroxinitrito (Vía-LAONP): ¿Protege o empeora el curso de la enfermedad de Chagas? María Eugenia Cárdenas, MSc1, Diego Torres, MD, MSc, PhD1,2, Ana María Mujica1, Jasson Sebastián Sanabria1 1Faculty of Health Sciences, Medical Program, Universidad Autónoma
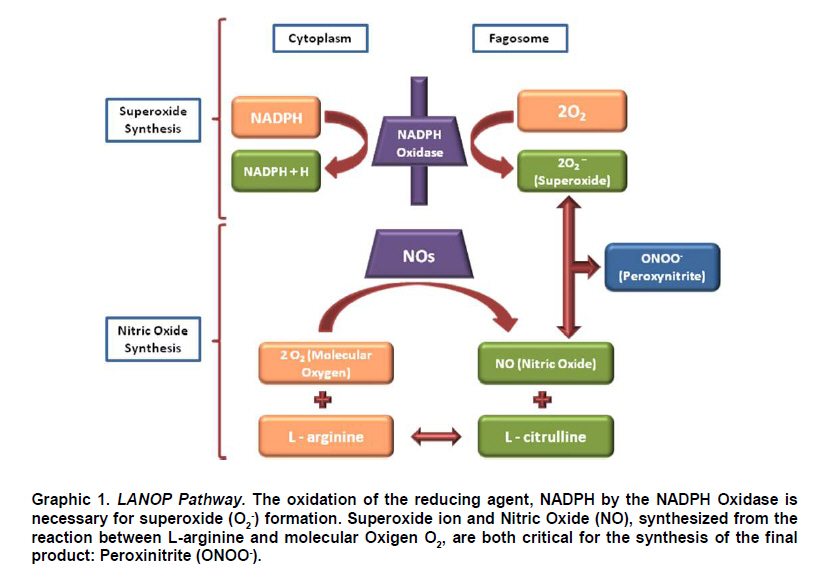
de Bucaramanga, Floridablanca, Colombia. Received for publication November 4, 2009 Code Number: rc10055 SUMMARY Many different immunological processes have been described in the Chagas infection, some of them associated with the Chagas disease. In this scenario, the L-Arginine-Nitric oxide (NO) –Peroxynitrite (NOOO-) pathway (LANOP pathway) appears as an essential component of that process. The relationship is well known between cytokines that can induce Oxide Nitric Synthase (iNOS) genes, such as TNF-α and IFN-γ, and other molecules that can inhibit their expression (TGF-β, IL-10 and others), which are involved in both acute and chronic stages of the disease pathogenesis. However the participation of the LANOP pathway seems complex, given that evidence shows different roles for it during the course of the infection. In this article, the authors review the immunological and inflammatory response leading to the activation of the LANOP pathway during the Chagas infection, and the role this via plays, including different effects, protector or deleterious, observed in parallel during the development of the infection. Keywords: iNOS; Nitric Oxide; Trypanosoma cruzi; Chagas disease; Chagasic cardiomiopathy; Peroxynitrite. RESUMEN En la infección chagásica se han descrito diversos procesos inmunológicos, algunos de los cuales se han asociado con la enfermedad. En este escenario, aparece la vía L-Arginina-Óxido Nítrico (NO) - Peroxinitrito (ONOO-) (Vía-LAONP) como un componente esencial de estos procesos. Se conoce la asociación entre citocinas inductoras de los genes de la Sintasa del Óxido Nítrico (iNOS) tales como TNF-α e IFN-γ, así como moléculas que inhiben su expresión (TGF-β e IL-10 entre otras), involucradas en la patogénesis tanto de la fase aguda como crónica de la enfermedad. No obstante, la participación de la vía-LAONP parece ser compleja, una vez que las evidencias señalan papeles diferentes de ésta durante el curso de la infección. Por tanto, los autores revisan la respuesta inmunológica e inflamatoria que da lugar a la activación de la vía-LAONP durante la infección chagásica, y el papel que ésta desempeña, incluyendo efectos diversos, tanto protectores como deletéreos, que han sido observados en paralelo durante el curso de la misma. Palabras clave: iNOS; Óxido nítrico; Peroxinitrito; Trypanosoma cruzi; Enfermedad de Chagas; Cardiomiopatía chagásica. The hemoflagellate Trypanosoma cruzi protozoan is the etiological agent of Chagas disease in Latin America, infecting close to 15-million people, even when there are nearly 28-million individuals exposed1. Its manifestations during the acute phase are non-specific. Two to three decades after primary infection, between 30 and 40% of the seropositive individuals in undetermined phase, present signs and symptoms of dilated cardiomyopathy2. Herein, we reviewed physio-pathological aspects of the Chagasic infection, with emphasis on the L-Arginine-Nitric oxide (NO) –Peroxynitrite (NOOO-) pathway (LANOP pathway), along with the most notable evidence of the dual actions (beneficial and deleterious) of these free radicals on the affected tissue. It is worth knowing and understanding the dimension acquired by the LANOP pathway in the physiopathology of the disease for the basis of future studies. INVASION OF THE HOST BY TRYPANOSOMA CRUZI AND INTERACTION WITH THE IMMUNE SYSTEMThe interaction between cells from different hosts and the parasite, a process of fundamental importance in understanding its pathogenicity, has been extensively reviewed by different authors3,4, and although it is beyond the objective of this review, we will address some relevant aspects given its close relationship with LANOP pathway. T. cruzi, an obligate intracellular protozoan with high genetic variability (lineage) within its sole species5, manifests a diversity of biological behaviors and cellular interactions, which require it to put into action its virulence factors, in response to the defense mechanisms of the host to be able to perpetuate its replication6-8. The parasite has two phases in its development cycle: one in the vertebrate host (which includes humans), and another in the insect vector. When the T. cruzi penetrates the vertebrate host, it multiplies locally as a amastigote, and appears in peripheral blood as a blood trypomastigote, responsible for parasitemia. From there, it is ingested by the triatomine, goes through the epimastigote stage, and reaches the hindgut of the vector, where it is multiplied as a metacyclic trypomastigote and is eliminated in the feces; this way, it becomes infectious to mammals. Here, the parasite penetrates via continuity solutions in skin or mucous membranes, thus closing its life cycle9. Many types of mammalian cells, including humans, have been studied as a model to understand the initial membrane-membrane interactions between host and parasite, through cell culture systems, cell lines, and primary cultures10,11. Some of the models mostly used by researchers have been mammalian cells, usually macrophages, fibroblasts, epithelial and muscle cells of murine origin, or cells from the phagocytic mononuclear system, proceeding from human peripheral blood12-14. It is known that in vertebrates the first contact of the T. cruzi occurs with the mononuclear fagocytes15,16, via Toll-like receptors (TLRs), and the recently described NOD-like receptor (NLRs), which recognize multiple molecular patterns from different pathogens17,18. There are other interactions between both cell surfaces, via lipid and protein sialoglyco-conjugate molecules, present in the mononuclear phagocyte and in the parasite, permitting cell activation through specific signals19,20. Among these molecules some lectins and integrins are described, as well as other molecules of cellular and vascular adhesion: fibronectin and sialic acid. The parasite manages to transfer itself through the phagocyte membrane, given that it expresses trans-sialidase-neuraminidase enzyme activity, and cysteine protease (Cruzipain)21,22. Other cells implied in the initial control, via induction of the iNOS expression, are the NK23. As of this moment, a series of complex and critical processes are generated to mediate the production of inflammatory mediators, initially at the expense of innate mechanisms of the immune system and, thereafter, these go on to guiding the actions of the antigen-presenting cells (APCs), which would be key for inducing the adaptive immune mechanisms24,25; two moments that could correspond in vivo to the acute and chronic forms of the infection. Within this context, NO, as a parasiticide molecule, is one of the main initial mediators relevant in parasite control; it is produced in considerable levels a few hours after activation of via TLR and NLR phagocytes, which induce the expression of the iNOS gene14,17,26. Given that some inflammatory mediators (Tumoral Necrosis Factor alpha, TNF-α, Interleukin 1, IL-1, and Interferon gamma, IFN-γ), also strongly induce the gene in various cell types, among them the mononuclear phagocyte; others like the transforming growth factor beta (TGF-β) can repress it11,27. NO is considered indispensable for infection control, at least in mice11,27-29. Hence, it is probable that its level of production, as well the equilibrium in the level of inflammatory mediators controlling said production, are key for the outcome of acute infection, as has been described30,31. The adaptive immune system is activated once the APCs process and introduce parasite antigens into T cells within the context of the HLA-II molecules. As in other intracellular infections, the Th1 profile would be responsible for greater IFN-γ production, which stimulates the production of NO from L-Arginine, by the activated macrophages, for the purpose of destroying parasites13,32,33; however, for several reasons, this response may be modified in favor of the T. cruzi, permitting perpetuation of the infection. In this sense, there is experimental description of various mechanisms of evasion or modification by the T. cruzi to defense mechanisms of the host, both in the innate as in the adaptive immunity. For example, the glycoprotein gp160, which has the trypomastigote, bonds to the C3b and inhibits recruitment of subsequent members of complement cascade, preventing the formation of the convertase and parasite lysis34, while modulating functions during the antigen presentation, like inhibiting the expression of HLA-II35. It has also been described to induce cell apoptosis of T cells (more Th1 than Th2), and diminishes the expression of IL-2 and IL-1231, which generates disequilibrium at the Th1/Th2 level, favoring parasitic persistence32,36. It may be said, then, that NO plays an important role in infection persistence and that in the parasite-host relationship there are specific aspects of the pathogen (sustained expression of surface glycoproteins that maintain the parasite-cell interaction, and capacity to evade the defense of the host), and specific aspects of the host (proper recognition, presentation and activation of immune mechanisms associated to parasite control), which determine the outcome of said infection37. L-ARGININE-NITRIC OXIDE-PEROXYNITRITE PATHWAYChemically, NO is a low-molecular weight radical gas (30 daltons), soluble in water and lipids. It reacts in water with the oxygen and its intermediary reactives14,38 to produce other radicals and anions of diverse stability. Given its characteristics, it easily spreads the eukaryote and prokaryote cell membranes. Once it is produced by any cell type, the human body performs its actions, physiological or physio-pathological, according to its synthesis triggering process. Because NO is produced in constitutive or inducible manner, according to the expression of the gene of the enzyme synthesizing it, this gas will have a different level and synthesis duration, but always with a short mean biological lifespan of a few seconds39 (6-10 s). The biochemical basis of the toxicity induced by the nitric oxide on phagocytized organisms, apparently depends on the combination of such with molecules containing iron in their structure, e.g., key enzymes in the cell respiration and replication cycle14 like the hapta-protein enzyme complex of the NADPH oxidase, critical in multiple cell processes. Not only does the phagocytic mononuclear system produce large and sustained amounts of NO under induction stimulus from iNOS, but the activity of this enzyme has also been detected in other cells like: hepatocytes, vascular smooth muscle cells, polymorphonuclear neutrophils and endothelium, among others. Hence, NO is involved in acute and chronic inflammation, as well as in the innate and acquired i mmunity39,40. Specifically, for the case of American trypanosomiasis NO has been described as a participating molecule in the pathogenesis of the disease, but with different effects in the diverse phases of the disease37. Also, NO has been described as key in the synthesis of several agents considerably important in the physiopathology of acute and chronic inflammatory processes among which there is peroxynitrite (ONOO-), which results from the reaction between NO and the superoxide ion (O2-). O2-, also described as a direct microbicide agent, is released from the granulocyte and macrophage respiratory chain, through a redox reaction in which the NADPH oxidase transfers an electron from the equivalent NADPH reducer to the molecular oxygen41 (Graphic 1). Although, initially NADPH oxidase was only described in phagocytic cells, currently it has also been identified in non-phagocytic cells like fibroblasts, endothelial cells, vascular smooth muscle cells, renal mesangial cells, and renal tubular cells. O2- is also produced during autoxidation of hemoglobin, myoglobin, and cytochrome c; as well as enzymes like xanthine oxidase, aldehyde oxidase, and a variety of flavin dehydrogenases41. The resulting superoxide is subsequently converted to various intermediate reactive species with microbicide functions consisting of structural damage on the microorganisms41. ONOO- is recognized as an oxidizing agent of highly reactive nitration, with a half-life of approximately 1 second at 37p C, pH=7.4, and stable in alkaline solutions41. It reacts with a variety of biomolecules, including lipids, proteins, carbohydrates, and deoxyribonucleic acid. From the point of view of physiopathological effects, ONOO- has been identified as an important microbicide agent, triggering lipid pero-xidation, sulfhydryl oxidation, inactivation of sodium transport, and nitration of tyrosine residues in a significant variety of proteins that include the inactivation of enzymes and/or receptors; associated to oxidative stress and consequential endothelial42. NITRIC OXIDE (NO) SYNTHESIZING ENZIMES The L-arginine amino acid was described in 1988 as the precursor in biosynthesis of nitric acid in endothelial cells. Said synthesis is catalyzed by a perfectly studied, classified, and characterized enzyme complex called nitric oxide synthase (NOS) of which there are three described isoforms, whose expression differs from one tissue to another, regarding the control of the gene transcription and the enzymatic activity. One is neural (nNOS), another is inducible (iNOS), and the other is endothelial (eNOS), or types I, II, and III, respectively14,38,43. The NO synthases have relevant differences in relation to inducer mechanisms of their expression, amounts, and duration time of the NO produced; thus, eNOS and nNOS, being of constitutive expression, depend on calcium/calmodulin for their activation and synthesize NO in picomolar amounts and for short periods (seconds to minutes), while iNOS is independent of calcium and is activated by another series of mediators that induce the gene expression to produce sustained nanomolar amounts of NO38. The catalytic reaction of molecular oxygen with L-arginine to form its two end products: nitric oxide and L-citrulline, is a reaction common to the three isoforms. Structurally, these enzymes are hemoproteins 40% similar to cytochrome P-450. They exist as dimers to be functional, have a reducer end and the other is oxidizing in each monomer, and in their active form bind molecules of NADPH, FAD, FMN, calmodulin, heme, and tetrahydrobiopterin38,44. PHYSIOPATHOLOGY OF THE CHAGAS DISEASE: PARTICIPATION OF THE LANOP PATHWAY IN THE IMMUNO-INFLAMMATORY RESPONSE The parasite starts the infection by penetrating as a metacyclic trypomastigote into the vertebrate host, and once it manages to invade a broad variety of cells, immunity is triggered in the host with active participation of multiple effector mechanisms that mediate the control of acute infection, permitting in some cases the progress to chronicity and predominantly affecting muscle tissue45. Nitric oxide (NO) is among these mediators, derived from iNOS, which for over a decade has been known for its great in vitro microbicide power, including Trypanosoma cruzi29. This parasiticide effect on trypanosomiasis is achieved via oxidizing mechanisms in response to the stimulation of the enzyme’s inducible gene, present in a large number of the body’s cells. In spite of the parasiticide effect of NO on T. cruzi29, some results have been found indicating that the participation of NO is not totally essential for this control46 and, furthermore, that to a certain extent -according to the state of the infection- it may play a predominant deleterious role in the pathogenesis of the chronic disease47. iNOS is mainly implied in the immunological response to respond against the Trypanosoma. Its gene is located in the human chromosome 17 and was initially described in the macrophage, although it has currently been found in different tissue, including cardiac muscular tissue. Genetic polymorphisms have been described within several loci of the gene codifying for iNOS; however, there is no evidence of association among these with the susceptibility for T. cruzi infection T. cruzi48. In non-stimulated macrophages, enzyme activity is not normally present; after a few hours of cell activation, through bacterial products (Lipopolyssacharide bacteria or LPS) or via pro-inflammatory cytokines28,49 (IL-1, IFN-γ, FNT-α), there is expression for synthesizing iNOS de novo. Once the gene is expressed, NO is produced for a long period (48-72 hours) and in great quantities, which permits the action of NO in the immunological response. Nevertheless, this may result in toxic manifestations for adjacent tissue and even for the tissue producing it, contributing to the pathogenesis of the disease, as in the case of experimental Chagas12. The same way iNOS can be stimulated, it may also be blocked by different inhibitor-type substances competing with L-arginine, such as L-N-monometilarginine (L-NMMA), NG-nitro-L-arginine methyl ester (L-NAME), and aminoguanidine43, which have demonstrated, via experimental studies, the protective effect of NO against infection with T. cruzi, given that parasitosis cannot be controlled in the absence of NO, or when it is inhibited50. It is worth noting the existence of a counter regulation form among the expression of such, like for example, NO produced in large amounts because of the stimulus of TNF-α on iNOS is countered since it inhibits to a certain extent the constitutive enzyme. Once the parasite is interiorized, its death depends on the production of NO. Said production by the activated macrophage is stimulated by TNF-α and IFN-γ, and by chemically attracting molecules like the platelet activating factor (PAF), C-C chemokines, and leukotriene B4, which it appears also directly induce the expression of iNOS51. Molecules like the macrophage colony stimulating factor (GM-CSF) may indirectly amplify the parasiticide effect of NO, inducing increased production of TNF-α49 and IL-12; thereby, favoring the synthesis of IFN-γ. Conversely, cells like NK cells (natural killer cells), present in the infection’s physio-pathological process, manage a direct amplifying effect of NO via induction of iNOS by the INF-γ2. Other specific-immunity molecules, like TGF-β and IL-10, also produced during Chagas infection, are negative regulators of NO production, revised in27, suggesting that the pathogenesis of the disease (at least in Chagasic cardiomyopathy observed in mice), depends on a fine balance between pro- and anti-inflammatory cytokines, i.e., cytokines from clones of CD4+ Th1 lymphocytes, but not those produced by clones of CD4+Th2 lymphocytes, activate the macrophages to eliminate the parasite with NO mediation13,32. Another interesting aspect of the trypanocidal response mediated by NO, confirmed experimentally by using iNOS-gene deficient mice, highly susceptible to infection, lies in the fact that the participation of the radical gas is apparently dependent on the elapsed time of infection. Thus, as noted in mice, NO is essential during the initial periods of the acute phase to decrease parasitemia and diminish mortality, but it is not equally relevant during the late period of the acute phase, or during the chronic phase. This reveals fundamental differences in the infection’s control mechanisms, which probably behave differently against other related microorganisms37. It was mentioned previously that one of the ways NO exerts its parasiticidal action is by binding to iron-containing structures, nevertheless, it is worth noting that when NO reacts with O-2 (equally synthesized by the macrophage), to produce ONOO- (more cytotoxic), it may be considered a positive modulator of the organism’s defense system. In other words, NO confers resistance to the macrophage activated with IFN-ã in the presence or absence of the metabolic burst, which was previously described53. Also, it has been shown that it could be involved in processes suppressing the immunological or immune-modulation system, to the extent that it induces in vivo and in vitro apoptosis of the cells affected during the acute phase of the experimental infection54. The apoptosis induced by NO involves the Fas-FasL interaction, whose inhibition (by using Fas expression deficient mice), generates a decrease in NO production as a consequence of increased synthesis of Th2 cytokines; which would attribute NO a regulating or modulating effect of the response through this mechanism55, as in the case of immune suppression against T. cruzi, via the apoptosis induction in T cells T54. Regarding the immunoregulatory effect mediated by NO, there is knowledge of a protein released by the T. cruzi parasite (Tc52), which synergizes with IFN-γ to produce NO through iNOS induction and, indirectly, stimulates synthesis of IL-12 and IL-1. The experimental immunization with this antigen partially improves the immune-suppressing state described during acute Chagas infection. However, given that immune response has only been detected against Tc52 in T. cruzi infection, it is probable that this parasitic molecule plays a role in modulating the biological functions of NO7. Until now, discussion has centered mainly on the trypanocidal effect of NO. However, studies of experimental Chagas in iNOS-deficient mice reveal parasitemia and mortality equal to the wild controls, suggesting that iNOS is not essential to control the infection46. Interestingly, studies conducted with mice immunized with T. rangeli and then infected with T. cruzi revealed protection and increased survival associated with the rise of the Th2 pattern and diminished Th1 pattern (including NO), which particularly questions the protective role of NO during the acute phase of the infection with T. cruzi56. Furthermore, it has been observed that nitric oxide could be responsible for the reactivity decrease of the Th1 immune profile57, which generates Th1/Th2 disequilibrium36. Nevertheless, it has also been noted that in animals infected with T. cruzi, there is a decrease of the Th1 pattern via NO-induced apoptosis, favoring the persistence of the parasite58. It must be noted that the increased levels of NO and TNF-α in chronic Chagasic individuals, correlated to low levels of antioxidants like glutathione peroxidase and superoxide dismutase, favor the evolution of the disease in chronically infected patients59. Additionally, other studies have shown that NO from iNOS plays an important role in the development and progression of ventricular dilatation and systolic dysfunction in acute Chagasic myocarditis observed in mice infected with T. cruzi60. CONCLUSIONS In summary, this review permits concluding that the LANOP pathway is involved in the pathogenesis of acute and chronic Chagas disease studied in experimental models and in infected patients. Nonetheless, the role of this pathway is not unidirectional, given that the actions of such are dependent on time, because depending on the acute or chronic stage of the disease process, the pathway may have different actions or even contrary, protective or deleterious. This situation challenges researchers into broadening and delving deeper into these aspects and clearly defining the true dimensions of the role of the LANOP pathway in the physio-pathological process of the trypanosomiasis and, consequently, its plausible pharmacological manipulation. Also, macrophages are definitely key cells in all aspects of host-parasite interaction occurring during infection; and when observing the diversity of responses these cells have against the very parasite and/or against cytokines produced during the course of the infection, it is easy to understand that a greater number of studies is rendered to understand the complexity of cell interactions taking place during different phases of the disease and, thus, be able to obtain greater clarity on the protective or deleterious role of NO against T. cruzi, given that frequently both effects have been observed during the infection. ACKNOWLEDGMENTS This review was conducted as part of the research project executed at Universidad Autónoma de Bucaramanga through financial support from Instituto Colombiano de Ciencia y Tecnología (COLCIENCIAS), contract N° 133-2000. Conflict of interest. None of the authors has conflicts of interest related to this study. REFERENCES
The following images related to this document are available:Photo images[rc10055f1.jpg] |
| |||||||||
{kind=link}