 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
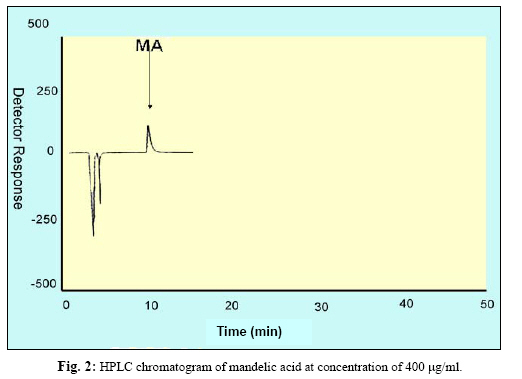
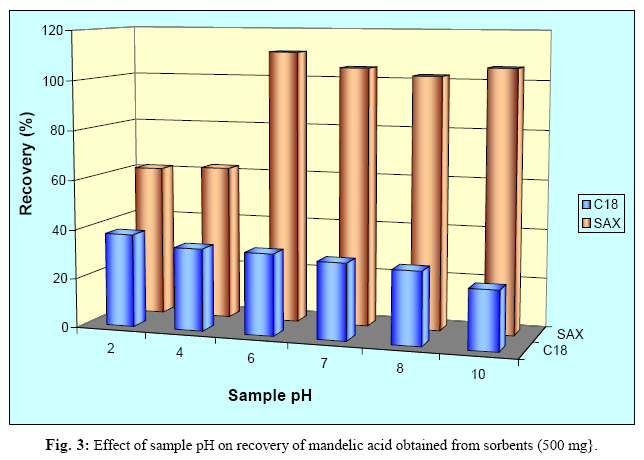
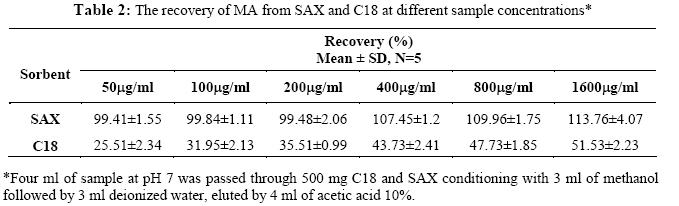
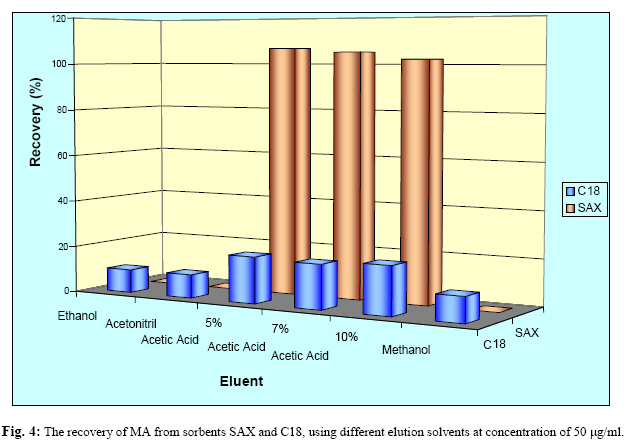
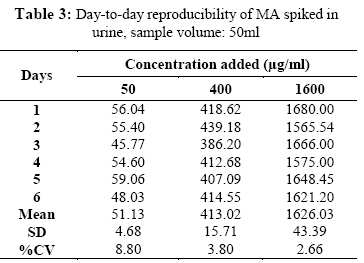
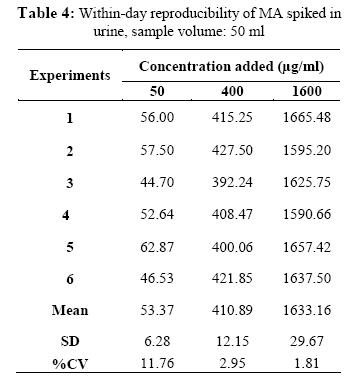
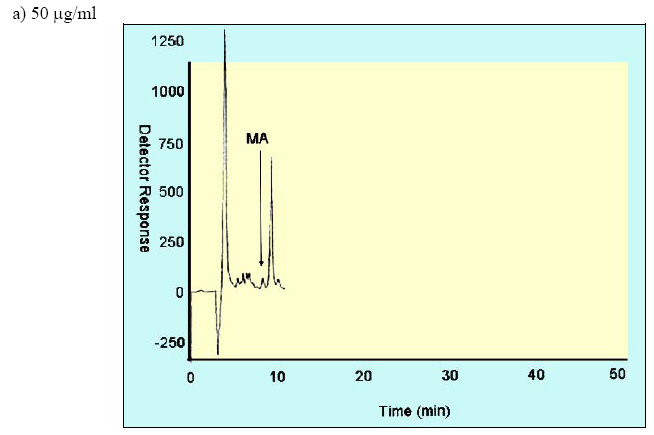
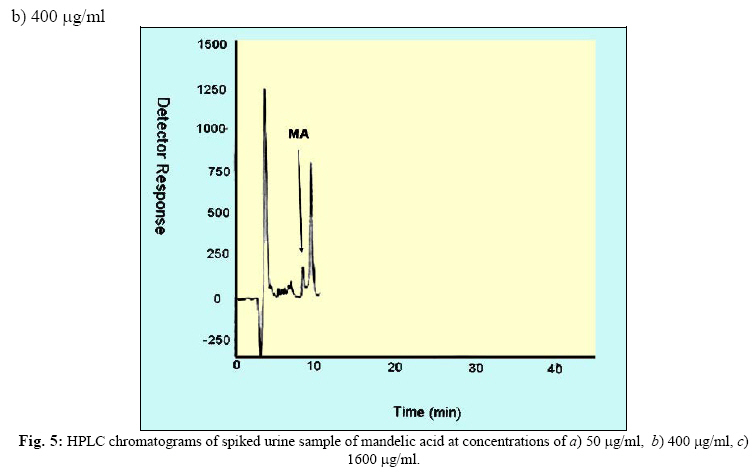
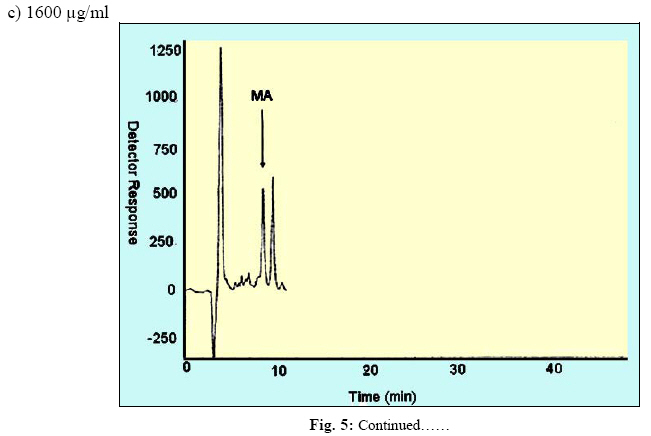
Iranian Journal of Environmental Health Science & Engineering, Vol. 1, No. 2, 2004, pp. 70-80 Optimization of SPE for Analysis of Mandelic Acid as a Biomarker of Exposure to Ethyl Benzene *S J Shahtaheri 1, M Abdollahi 1, F Golbabaei 1, A Rahimi-Froushani 2, F Ghamari 1 1Dept. of Occupational Health, 2Dept. of Epidemiology and Biostatistics, School of Public Health, Tehran University of Medical Sciences, Iran *Corresponding author: E-mail: sjtaheri@sphtums.com Tel: +98 21 88951390, Fax: +98 21 66462267 Code Number: se04019 ABSTRACT Ethyl benzene is an important constituent of widely used solvents in industries and laboratories, causing widespread environmental and industrial pollutions. For evaluation of occupational exposure to such pollutants, biological monitoring is an essential process, in which, preparation of environmental and biological samples is one of the most time-consuming and error-prone aspects prior to chromatographic techniques. The use of solid-phase extraction (SPE) has been grown and is a fertile technique of sample preparation as it provides better results than those of liquid-liquid extraction (LLE). In this study, SPE using bonded silica has been optimized with regard to sample pH, sample concentration, elution solvent, elution volume, sorbent type, and sorbent mass. Through experimental evaluation, a strong anion exchange silica cartridge (SAX) has been found successful in simplifying sample preparation. The present approach proved that, mandelic acid could be retained on SAX sorbent based on specific interaction. Further study was employed using 10% acetic acid to extract the analyte from spiked urine and gave a clean sample for HPLC-UV system. In this study, a high performance liquid chromatography, using reverse-phase column was used. The isocratic run was done at a constant flow rate of 0.85 ml/min, the mobile phase was water/methanol/acetic acid and a UV detector was used, setting at 225 nm. At the developed conditions the extraction recovery was exceeded 98%. The factors were evaluated statically and also validated with three different pools of spiked urine samples and showed a good reproducibility over six consecutive days as well as six within-day experiments. Keywords: Sample preparation, Solid phase extraction, Biological monitoring, Chromatography INTRODUCTION Due to increasing concern about toxic substances such as ethyl benzene in the environment and workplaces, it is becoming more important to monitor such chemicals and their metabolites in order to evaluate risk hazards and potential problems caused by exposure to toxic compounds (Keymelulen et al., 2001; Sperlingova et al., 2003). Ethyl benzene is an important industrial chemical because of its occurrence in mineral oil and its formation in many combustion processes, causing widespread environmental and industrial pollutions (Keymelulen et al., 2001). Although mandelic acid (MA) was identified as a urinary metabolite long time ago, it is now going to be a more popular biomarker for occupational and environmental exposure to ethyl benzene. In biological matrices, either exposed compounds or their metabolites mostly are present at a trace level, causing major problems in their determination stages. (McDowall, 1989; Shahtaheri et al., 1995, 1998). Therefore, an essential need for sensitive and selective techniques for the analysis of trace chemicals in environmental and biological matrices have been clearly recognized (McDowall, 1989; Poole et al., 1990, Hennion et al., 1993; Maria 2000). Although the use of detection system has improved the selectivity of the analytical procedures, these sensitive and selective methods require extensive equipments; moreover, they may not be available in most laboratories. Consequently, sample pre-treatment procedures which can be performed in any laboratory have been developed to simplify analytical approaches as these methods reduce expenses too (Barcelo 1991; Shahtaheri, Stevenson 2001). Although derivatization reactions performed either before or after analytical techniques, can enhance the sensitivity of the assay, this extra performance is not often a favorite stage in sample preparation followed by analysis. Although many analytical methods still use liquidliquid extraction (LLE) to perform sample clean-up (Ibrahim et al., 1988; Liu et al., 2001), in this procedure, large volume of solvents, having undesirable environmental concerns are used as well as problems associated with the technique to be automated. In addition, the recovery obtained from LLE is not often suitable and reproducible. In contrary, solidphase extraction (SPE) methods using silica or bonded silica have proven useful in simplifying sample preparation prior to HPLC-UV. Isolation and purification of the compound of interest can be achieved in a short time and only low volumes of` solvents are used during the application of the method. The use of commercially available low cost vacuum manifolds allows many samples to be processed simultaneously. Furthermore, complete automation of procedures based on SPE is now possible using commercially available instrumentation (Focant et al., 2004; Pettersson et al., 2004). A wide range of phases from many suppliers based on silicas are also available including reversed phase, normal phase, ion exchange and mixedmode phases. The phases can be screened and selected, depending on chemical nature of the analyte (Hennion 1999), therefore, the variety of available phases can improve the selectivity of sample preparation method. This study was aimed to achieve the optimum factors necessary for development of an optimized procedure for MA (Fig. 1), leading to a simple protocol of solid phase extraction method (Coline et al., 2000). MATERIALS AND METHODS Reagents Mandelic acid (99%) (MA) as standard was obtained from Merck, Germany. Methanol, ethanol, acetonitril, and acetic acid (all HPLC grade), deionized water, and standard buffered solution at three pH values (4.00±0.02, 7.00±0.02, and 9.00±0.02) were also purchased from Merck, Germany. Octadecyl (C18) and strong anion exchange (SAX) sorbents (100 mg and 500 mg) were obtained from Macherey-Nagel, Germany and used for solid phase extraction procedure. Apparatus A Vac-Elute Vacuum Elution System was used for retention and elution of silica cartridges. A digital pH meter from Hanna, Singapore was used for pH adjustment. The amount of reagents was measured, using a Satorius balance for milligram quantities or less. Quantitative liquid transfers were performed with pipette (Socorex, Germany). The HPLC apparatus used in this study included the following equipments: a K-1001 single piston pump (Knauer, Germany), the analytical column was a C18 (25 cm × 4.6 mm i.d., 5 μm) purchased from Hichrom Limited, Reading, UK. The detector was a K-2600 LC-UV spectrometer obtained from Wellchrom, Germany. The system was linked with a LaserJet 1200 series printer for recording the chromatograms, using a 1456-1 Chromogate Data System, Version 2.55 (Knauer, Germany). Because the reagents used in this study were HPLC grade, there was no need to filter them. However, the analytical column in HPLC system was equipped with a filter on the top. Solvents and mobile phase used in HPLC analysis were degassed by an on-line degasser attached to the solvent delivery system. Optimized sample preparation procedure In this study, SPE using bonded silica including SAX and C18 (100 mg and 500 mg) has been optimized with regard to sample pH, sample concentration, elution solvent, elution volume, sorbent type, and sorbent mass. The cartridges were conditioned with 3 ml of methanol followed by 3 ml HPLC water. Care was taken to prevent the cartridges from drying. The samples were then passed through the columns at a flow-rate of 1-2 ml/min. The cartridges were then washed with 3 ml of different solvents. Finally, the MA was eluted from the column with 4 ml different solvents. The extracts were then analyzed by HPLC-UV. Chromatographic conditions The pump was operated at 0.85 ml/min, UV detection wavelength was set at 225 nm, the mobile phase consisted of water/methanol/acetic acid, 69:30:1 (v/v/v), flow rate was 0.85 ml/min, injection volume was 100 µl, the analytical column was C18 (25 cm × 4.6 mm i.d., 5 μm), and the ambient temperature was used for the chromatographic system. Under these conditions, MA was eluted and detected in around 10 minutes. In this study, peak area was used as detector response and extraction recoveries were calculated by comparison of the peak area in the chromatogram of extracts with those in the chromatogram of standard solutions prepared in the same solvent as following: Recovery (%) = Peak area (sample)/peak area (standard) × 100 More experiments were performed on spiked urine to valid the present method. Spiked urine can be a suitable model as it may contain interfering constituents similar to real sample (Laurens, et al., 2002). The spiked samples of 50 ml of MA were used for extraction followed by HPLC-UV determination. Linear standard curve (for extracted samples) over the range 50-1600 μg/ml were obtained each day (n=6) with correlation coefficient of 0.999 or greater. The extraction procedure was reliable and reproducible from day-to-day and within-day. RESULTS In order to achieve the optimum chromatographic conditions for analysis of mandelic acid, variables including mobile phase composition, UV wavelength, injection volume, and mobile phase flow-rate were optimized. Fig. 2 shows chromatogram of MA detected at around 10 min. In order to optimize SPE, there were several factors by which retention and elution could be altered. First, sorbents including C18 and SAX, containing 100 and 500 mg of bonded silica were evaluated for extraction recovery of MA. After conditioning the columns with 3 ml methanol followed by the same volume of deionized water, 4 ml of MA standard at different concentrations were applied. Retained analyte was washed with 6 ml of acetic acid 1% followed by elution with 4 ml acetic acid 10%. Table 1 shows the recovery of mandelic acid obtained from two mentioned cartridges. The 500 mg C18 and SAX cartridges were activated and conditioned according to the method explained. Four ml of sample at different pH values of 2, 4, 6, 7, 8, and 10 were applied. The columns were then washed and retained analyte was eluted using the same procedure as explained beforehand. Fig. 3 shows the influence of sample pH on extraction recovery for MA. In order to evaluate the effect of sample concentration on SPE performance, different concentrations of MA ranged from 50 to 1600 μg/ml mentioned in Table 2, were prepared using deionized water. Another experiment performed during this study was evaluation of the eluent strength on MA recovery. Six solvents were screened for their ability to produce optimum elution of the retained MA from the C18 and SAX sorbents. They were ethanol, acetonitril, acetic acid 5%, acetic acid 7%, acetic acid 10% and methanol. The same sequence of conditioning, washing, and elution was used as in previous section. The results of this process are shown in Fig 4. Tables 3 and 4 show the results obtained in method reproducibility experiments. Also, Fig. 5 (a, b, c) shows the HPLC chromatogram of spiked urine sample of MA at concentrations of 50, 400, and 1600 μg/ml. DISCUSSION Analytical column widely used for analysis of such compound is generally reversed phase, in which, C18 was preferred due to its frequent use and efficient results in the trace analysis of organic acids (Shui and Leong, 2002; Tormo and Izco, 2004). The wavelength of 225 nm was more sensitive for determination of the analyte. In this study, the flow-rate of the mobile phase was also screened, in which, 0.85 ml/min was a suitable flow-rate to get an optimum retention time for MA chromatogram. Using these conditions, the compound of interest was eluted in around 10 minutes as shown in Fig. 2. The retention time of mandelic acid can be changed by increasing different concentrations of organic modifier in the mobile phase. Therefore, retention time (k-value) can be varied by changing the composition of the mobile phase in order to isolate the analyte from interferences contained in the sample solution. From the result given in Table 1, it was deduced that, SAX cartridge was more satisfactory for efficient recovery of the MA. It seems that, the polarity of the sorbents as well as the hydrophlilicity of the compound can be the major factors for the mechanisms occurred. As can be seen, a little interaction was taken place with the C18. The quantity of the sorbent was then also screened, in which, it was indicated that, the greater quantity of the sorbent, the greater the sample breakthrough volume, and greater the elution solvent volume. Due to the type of interaction, providing and efficient recovery, SAX cartridge was selected for further optimization steps. The results showed that efficient recovery was obtained from SAX using sample pH 7 (Fig. 3). However, the amount of analyte recovered from SAX at sample pHs 6, 8, 10 were also efficient. From these pH values, sample pH of 7 was selected for further study as this value of pH seems to be rather mold. This investigation showed that the pH of the sample should be adjusted according to the chemistry of the compound of interest. MA is ionizable compound (pKa is 3.85) when the pH is 2 unites more than the pKa. Therefore, it was necessary to adjust the pH of the sample adequately in order to ionize MA completely and ensure that the compound was in appropriate ionic or weakly associated form to achieve efficient retention by the solid phase sorbent using ionic interaction mechanism. The extraction recoveries obtained from C18 at different pHs were still low, in which, the non-polar mechanism is the main reason of such phenomenon as MA is an ioizable solution. It also seems that, when the sample pH is adjusted at bellow pKa, C18 is not more stable, so, the compound cannot be retained on C18 adequately. Ideally, the extraction recovery should not be sample concentration dependent.In other words, for the method to be useful there should be no significant difference in recovery over the expected concentrations range of the compound to be analyzed. Table 2; gives the recoveries obtained after passing 4 ml sample at different sample concentrations followed by elution with 4 ml acetic acid 10%. As can be seen, the recoveries are independent of sample concentrations over the concentrations range studied. During this experiment, the breakthrough fraction was also analyzed and no breakthrough of the compound was detected. Although the limit of detection of the optimized procedure was 4 µg/ml, applying the high sample volume of 200 ml, concentration of 50 µg/ml was compatible to the biological exposure index (BEI) for evaluation of occupational exposure. Understanding the chemistry of the compound under analysis such as its hydrophilicity or ionizability can be useful in designing appropriate conditions to obtain efficient extraction recovery. Highly hydrophilic compounds result in strongly retained analyte making elution difficult and subsequent given poor recoveries from ionic sorbents. Although three different percentages of acetic acid can extract MA efficiently, acetic acid 10% seems to be an optimum eluent compare to other solvents to break completely hydrophilic interaction between sorbent and analyte of interest (see Fig. 4). According to the chemistry and solvent composition, it can increase the solubility of the analyte as well as minimizing the physical losses on sample handling. However, in case, other percentages (5%, 7%) of acetic acid can also be of preference considerations. Enrichment of the analyte in SPE is achieved by applying large volumes of sample and eluting the analyte in a minimum volume of eluent. The eluent volume must be just sufficient to elute the compound of interest from the sorbent. The result obtained from an evaluation of elution volume showed that the smallest satisfactory volume for acetic acid from 500 mg of sorbent, was 4 ml. As a consequence, the volume required to elute analyte from the sorbent, depends on two important parameters. First, the capacity factor (k') of the compound of interest, shows the strength of its retention. Solvent with grater elution strength can be used to elute an analyte in less volume but may incorporate undesirable contaminants into the eluted fraction, secondly, the sorbent mass used in SPE, in which,using a larger sorbent mass cartridges require an increased elution volume to be applied. Finally, the coefficient of variation (CV%) of 8.80, 3.80, and 2.66 were obtained for 50, 400, and 1600 μg/ml respectively for day-to-day and coefficient variations of 11.76, 2.95, and 1.81, at the same concentrations, respectively for within-day, showing suitable accuracy and precision for the optimized method (Table 3, 4). The detection limit of the method using spiked sample volume of 50 ml was 4 μg/ml as well as reproducible and quantitative recoveries, ranging from 97% to 100% were possible. Through this study factors influencing SPE were optimized, showing efficient sample preparation procedure for mandelic acid as a biomarker of ethyl benzene as solid phase extraction method using silica-bonded is more advantageous than liquid-liquid extraction. Depending on chemical and physical properties of the analyte, manipulating of the factors including sorbent type, sorbent mass, sample pH, sample concentration, eluent and elution volume can play a main role in optimizing the method, providing reliable, easy to use, and cost effective procedure to overcome difficulties associated with other sample preparation techniques. Applicability of the method for treatment of different classes of pollutants such as pesticides and different hydrocarbons can make the technique to be popular when a selective and sensitive trace residue analysis is required. The authors sure that, the SPE is a highly fertile area for sample preparation methods and based on the needs and facilities, these method protocols can be more developed in the near future. ACKNOWLEDGEMENTS This work has received official and financial support from the School of Public Health, Tehran University of Medical Sciences, Iran in order to support Mr. M Abdollahi. Sincere assistance of Center for Environmental Research (CER) of Tehran University of Medical Sciences (TUMS) is highly appreciated. REFERENCES
© 2004 Tehran University of Medical Sciences Publications The following images related to this document are available:Photo images[se04019t3.jpg] [se04019f5c.jpg] [se04019f5b.jpg] [se04019f5a.jpg] [se04019f1.jpg] [se04019f4.jpg] [se04019t2.jpg] [se04019t1.jpg] [se04019t4.jpg] [se04019f2.jpg] [se04019f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}