 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
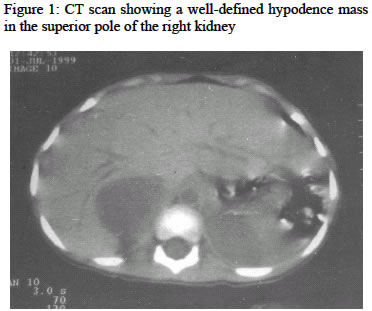
Nigerian Journal of Surgical Research, Vol. 6, No. 1-2, Jan-June, 2004, pp. 64-66 Case Report Phaeochromocytoma in a 4-year old girl: case report B. M. Gali, A. G. Madziga, *H. A. Nggada and **A. U. Hamid Departments of Surgery,
*Pathology and **Radiology, University of Maiduguri Teaching Hospital, Maiduguri Code Number: sr04019 ABSTRACT This is a report of a 4-year old female Nigerian patient with phaeochromocytoma, a rare surgically correctable cause of hypertension, highlighting the problems in diagnosis and management in our sub region with review of the literature. Key words: Phaeochromocytoma, diagnosis, management INTRODUCTION Phaeochromocytoma are tumors that are derived from chromophil cells of the adrenal medulla, which produces, stores and secrete catecholamines.1, 2 When the tumors arise from chromaffin tissue of the sympathetic ganglia in the thorax or abdomen they are known as paragangliomas or extra adrenal phaeochromocytoma.1 Phaeochromocytoma is a rare surgically correctable tumor that is found in only about 0.1-0.6% of patients with hypertention.1-3 The incidence among Africans is un known with few reported cases.4-9 Phaeochromocytoma exhibits a striking variety of presentation, thus its regarded as the “great mimic”. 2, 3 Because of the rarity and variability in clinical presentations, making a diagnosis is difficult especially in children with out high index of suspicion. Phaeochromocytoma is regarded as a 10% tumor, as it fits with the classic “rule of thumb” of 10% for bilaterality, extra-adrenal location, multicentricity, malignancy, or occurrences in a familial setting.1-3 Congenital narrowing (coarctation) of the aorta is a more frequent cause of hypertension in infants and children, however other cause of hypertension in children like renovascular hypertension needs to be excluded. We report this case of phaeochromocytoma in a 4-year old female Nigerian to highlight the problems with the diagnosis and management of this uncommon condition. CASE REPORT A 4-year old female Nigerian was admitted to the University of Maiduguri Teaching Hospital (UMTH) in June 1999 having visited several other hospitals with complains of weight loss despite voracious appetite, fever and headache, which started two years prior to the presentation. She also had a history of poor effort tolerance with occasional bluish discoloration of her lips, cough, polyuria, excessive sweating, heat intolerance and palpitation. No history of contact with a person with chronic cough. There was no family history suggestive of the disease. On account of the weight loss, fever and cough, she was treated for chest infection with Ampiclox, then subsequently for pulmonary Tuberculosis with no improvement. Patient was taken to another general hospital, where a diagnosis of congenital heart disease (hole in the heart) was entertained. She was then referred to the University of Maiduguri Teaching Hospital for further management. Examination revealed a young girl, small for age (weight 11.5kg), irritable, not pale, acynosed, and not jaundice. Heart rate 150 beats/minute, Blood Pressure on admission was 160/110 mmHg; first and second Heart sounds were heard with grade 3/6 pansystolic murmurs. Abdominal examination revealed a vaguely palpable right flank mass, firm and non tender. A diagnosis of neuroblastoma was initially made, but with the fluctuating high blood pressure, pheaochromocytoma was suspected. Abdominal ultrasound scan was then requested which revealed a mixed echogenic mass lesion at the superior pole of the right kidney measuring 4.7x4.7cm. Computerized tomography (C-T) scan confirmed the lesion to be a right adrenal tumor (Figure 1). Her 24 hours urinary vanillylmandelic acid (VMA) assay could not be done here, as the reagent for the test was not available then. The urine sample was sent to the University College Hospital (UCH) Ibadan for analysis that came with a high value of 60 mg/24hours (normal range <7.0mg/24hours). Chest X-ray and Electrocardiogram (ECG) showed features of left ventricular hypertrophy (LVH), packed cell volume was 33%, urea, electrolytes and creatinine were within normal limit and fasting blood glucose of 5.3mmol/L. Patient was commenced on intravenous hydrallazine 0.5mg/kg start then 0.25mg/kg slowly, hydrochlorothiazide 20mg 12 hourly, frusemide 10mg 12 hourly. The blood pressure remained consistently high with paroxysms up to210/160mmhg. In consultation with the anesthetists, the drugs were changed to propranolol 10mg 8hourly, phenoxybenzamine 2.5mg 12 hourly and labetolol 50mg 12 hourly. The dose of labetalol was later increased to 100mg; this lowered the blood pressure eventually to 160/100mmhg after two weeks of commencement of therapy. Phentolamine required for intraoperative use was not available locally; this delayed the surgery for another 2 weeks. Pre-operatively patient was well hydrated with 4.3% dextrose in 0.18% saline, evidence by the adequate hourly urinary output of 30ml/hour. She was given 250mg IV cefuroxime at induction of anesthesia. General anesthesia was used with muscle relaxation, pentazosine 10mg was given as intraoprative analgesia. Intraoperatively tumour mobilization was characterized by sudden increase in blood pressure up to220/160mmhg, which was managed with intravenous phentalomine 10mg slowly. Following removal of the tumour and during closure the blood pressure fell precipitously, and could not be restored with rapid infusion of 500mls of normal saline, 250mls of blood infusion and o.2mls adrenaline (1 in 1000) Injection and 50mg of hydrocortisone. The patient remained hypotensive and did not recover fully from anaesthesia. Her condition continued to deteriorate in the intensive care unit (ICU) and eventually died 36 hours postoperatively. Grossly the tumour was ovoid, cystic, grayish-orange measuring 5 x 3 x 3cm and weighing 70gm. Histology showed a well-defined typical cell nests (Zellballen) surrounded by delicate fibro vascular stroma, with moderate nuclear hyperchromasia and atypia in some areas. A delicate fibrous capsule that is partly surrounded by adrenal cortical tissue surrounds the tumour. The features are consistent with phaeochromocytoma. Consent was not given for postmortem examination. DISCUSSION Hypertension is a frequent cause of devastating illness and death. In 1981 the American Heart, Lung and Blood institute reported that one in six Americans (35,000,000), has definite high blood pressure, about half are aware and only about 14% are adequately treated.1 Surgically correctable hypertension accounts for 5-15% of the total spectrum of this disease. This includes, coarctation of the aorta, renovascular hypertension, primary hyperaldosteronism, hyperadrenocortism, and phaeochromocytoma. Coarctation of the aorta, a congenital narrowing of the Aorta is a more frequent cause of hypertension in infants and children. Diagnosis is most easily made comparing blood pressures in the upper and lower extremities. A difference of 20-40mmhg may be found. Older children and adults may exhibit over development of the upper body and under development of the lower limb. Renovascular hypertension is hypertension associated with renal artery occlusive disease. It is one of the common cause of hypertension in children and is usually sustained and severe and not easily controlled by medication. Primary hyperaldosteronism, is the increase in primary aldosterone secretion by the zona glomerulosa of the adrenal cortex, and is associated with high blood pressure, hypokalaemia, hypernatremia and hypervolaemia. Clinically a patient complains of muscle weakness, headache and malaise. Hyperadrenocortisism (Cushing’s disease) is characterized by hypertension, truncal obesity, striae, ammenorhoea and hirsuitism due to increased circulating glucorticoids. Women thirty to fifty years of age are most commonly affected. Phaeochromocytoma is found in only 0.1-0.6% of patients with hypertension, and with no sex predilection.1-3 The true incidence among Africans is unknown, based on the few isolated cases reported, it would seem to be rare. 4-9 To the best of our knowledge this would be the first report from the Northeastern part of Nigeria. Its incidence is higher with multiple endocrine neoplasia (MEN) type II and some other familial neurocutaneous syndromes, such as von Recklinghausens Neurofibromatosis and von Hippel-Lindau syndrome than in the general population.10, 11 Extra-adrenal phaeochromocytoma is reported to be more common in children than in adults and also more common in Africans.9 The diagnosis of phaeochromocytoma is based on high index of suspicion. Because of the variability of clinical manifestations the tumours have been regarded as the “great mimic”. Our patient presented with the most common symptoms of, palpitation, headache, fever, sweating, weight loss, tremors, polyphagia and increased blood pressure as a result of circulating catecholamines that increase small vessel tone. These common symptoms has been reported with similar frequencies by several other authors.1-5 The presence of cough and occasional bluish discoloration of the lips noted in this patient lead to the misdiagnosis and treatment for chest infection (pneumonia and tuberculosis) and diagnosis of a “hole in the heart” (congenital heart disease). Several other unusual presentations have been reported by Short, et-al12 where patients presented with severe obstinate constipation, Keller, et-al13 described a patient who presented with lactic acidosis, while Thomas, et-al14 described adrenergic crisis due to phaeochromocytoma. Delays in making diagnosis in this patient allowed the ravages of untreated hypertension for 24 months to cause the morbidity and mortality, because of the secondary involvement of the heart, brain and kidney. A large series of 55 cases, treated and followed up by Favia, 2 showed an age range of 10-63 years. Other reasons to suspect a surgically correctable lesion includes, sudden onset of severe or malignant hypertension, previously easily controllable hypertension that becomes labile or difficult to treat, or when there is a sign of hormonal excess. When a surgically correctable cause of hypertension like phaeochromocytoma is suspected, a screening investigation for increased 24-hour urinary vanillyl mandelic acid (VMA) is suggested as part of patient evaluation. However, recently serum VMA is said to be more sensitive as it detects smaller changes in catecholamine levels. Normal urinary 24 hours VMA is < 7mg/24hours and the normal venous serum levels is 1ug/L using the trihydroxyindole method.15 The screening examination should lead to more definitive localizing procedures that play a fundamental role in the management of phaeochromocytoma. Computed tomography (CT) scan and magnetic resonance imaging (MRI) are reported to have an accuracy of 85 to 95% and the sensitivity of 131/123I-metaiodobenzylguanidine (MIBG) scintigraphy ranges between 77% and 88% with a specificity of 88% to 100%. When the three procedures are combined, it yields an accuracy of 100%. 2, 3 Angiography and venous sampling techniques are reserved for tumours difficult to locate. While, transcatheter brush biopsy of inferior vena-caval tumour thrombi is a newer technique, but is said to has a limited applicability at this time.2 Preoperative treatment plays a major role in the care of patient with phaeochromocytoma. Optimum control of blood pressure is often difficult, as in this patient the blood pressure initially could not be controlled with hydrallazine, hydrochlorothiazide and propranolol, and even when labetolol, 50mg was increased to 100mg 8 hourly, the blood pressure still was not effectively controlled. However, the experience of Njoh, et-al 9 managing phaeochromocytoma is different, there was poor response on hydrallazine, hydrochlorothiazide and propranolol initially, but within 3 weeks on labetolol 100mg 8 hourly the blood pressure returned to normal. Phentolamine 10mg was used intraoperatively in controlling the hypertensive crisis that usually follows tumour handling. After removal of the tumour operatively, hypotension secondary to vasodilatation and hypovolaemia are known to occur.1 It is probable that, the prolonged preoperative use of antihypertensive drugs in high doses to control her hypertension may have contributed to the precipitous fall in the blood pressure following removal of the tumour. Because of the cardiovascular instability that occurs intraoperatively, it is necessary to place a pulmonary arterial wedge catheter (PAWC) or at least central venous pressure (CVP), where adequate volume replacement and cardiac status can be monitored in the intensive care unit (ICU) where the facilities exist. The surgical approach usually advocated is via transperitoneal or flank incision and currently laparoscopically.3 Transabdominal adrenalectomy has the added advantage of offering the opportunity to inspect the paraspinal area, bladder and the organ of Zuckerkandl to exclude the presence of extra-adrenal tumour. Phaeochromocytoma is rare, and high index of suspicion is required for early diagnosis. Timely surgical intervention and adequate therapeutic and monitoring facilities should keep the morbidity and mortality be low. REFERENCES
Copyright 2004 - Nigerian Journal of Surgical Research The following images related to this document are available:Photo images[sr04019f1.jpg] |
| |||||||||

{kind=link}