 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Tanzania Journal of Health Research, Vol. 13, No. 1, January, 2011, pp. 50-60 Detecting adenosine triphosphatase 6 (PfATP6) point mutations that may be associated with Plasmodium falciparum resistance to artemisinin: prevalence at baseline, before policy change in Uganda Erasmus Kamugisha1*, Hakim Sendagire2, Mark Kaddumukasa3, Nizar Enweji4, Fatemeh Gheysari4, Göte Swedberg4 and Fred Kironde2 1Department of
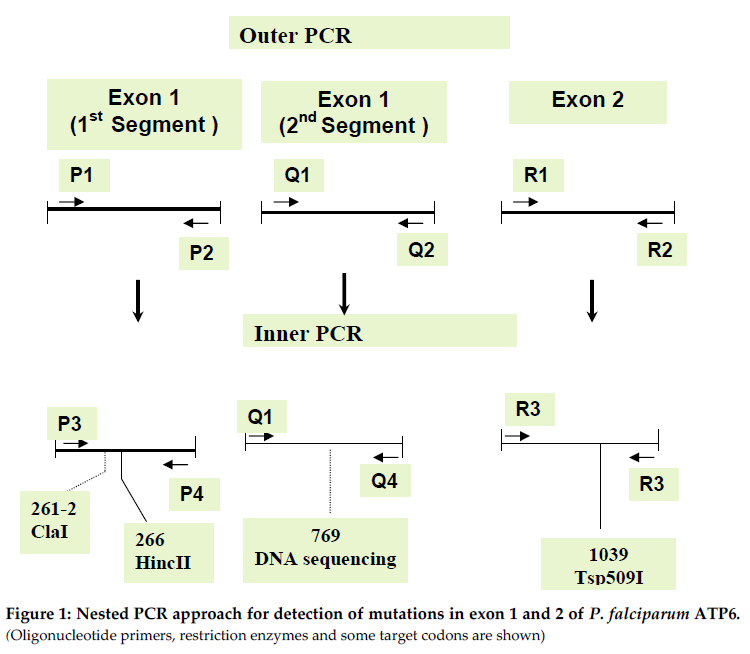
Biochemistry, Bugando University College of Health Sciences, Mwanza, Tanzania Code Number: th11007 Abstract The artemisinin based combination therapy (ACT) of artemether and lumefantrine (Co-artem) has recently replaced chloroquine and fansidar as the first line treatment policy drug in Uganda. It is necessary to develop practical procedures to monitor the likely emergence and spread of artemisinin resistant P. falciparum strains. We have analyzed the genotypes of PfATP6 in parasites from 300 stored filter paper samples from malaria patients who were diagnosed and treated in the years 1999 to 2004 at three field sites in Uganda. This is a period just prior to introduction of Co-artem. In order to develop a simple molecular procedure for mutation detection, regions of PfATP6 encoding protein domains important in artemisinin binding was amplified by nested PCR. Three DNA products, which together contain most of the coding region of amino acids located within the putative active site of pfATP6 were readily amplified. The amplified DNA was digested by restriction enzymes and the fragments sized by agarose gel electrophoresis. For the important codons 260, 263 and 769, methods using engineered restriction sites were employed. We did not find mutations at codons for the key residues Lys 260, Leu263, Gln266, Ser769 and Asn1039. Nucleotide sequencing of pfATPase6 gene DNA from at least 15 clinical isolates confirmed the above findings and suggested that mutations at these amino acid residues have not emerged in our study sites. Key words: PfATP5, artemisinin, resistance, SERCA, Plasmodium falciparum, Uganda Introduction Resistance to the cheap and widely available antimalarial drugs such as chloroquine (CQ) and sulfadoxine-pyrimethamine (SP) has increased rapidly in most parts of the world prompting National Malaria Control Programmes (NMCP) to recommend the use of Artemisinin based Combination Therapy (ACT). The Ugandan NMCP (UNMCP) recommended the combination of artemether and lumefantrine (Co-artem) as the first line drugs for treatment of uncomplicated malaria which was implemented in 2005. Co-artem replaced the hitherto recommended CQ+SP combination whose effectiveness was reduced substantially in the 5 years (2000-2005) when it was the recommended first line policy drug in Uganda. Artemisinin derivatives are highly attractive antimalarials because they act rapidly, are well tolerated, and are currently not limited by resistance. Despite that a high rate of recrudescence after artemesinin monotherapy (with its short half-life and other pharmacodynamic properties) has been reported (Giao et al., 2001; Ittarat et al., 2003). There is also evidence of delayed parasite clearance in Western Cambodia (Dondorp et al., 2009), which could be interpreted as signs of emerging resistance in the region (Carara et al., 2009; Maude et al., 2009; Rogers et al., 2009). In other parts of the world including Africa it is still effective when given in adequate dose regimens in combinations with longer-acting antimalarials such as mefloquine or lumefantrine (Sinclair et al., 2009; Byakika-Kibwika et al., 2010) The proper mechanism of action of artemisinins is still debatable (O’Neill et al., 2010; Witkowski et al., 2010). It has been proposed that the artemisinins work by binding and inhibiting P. falciparum ATPase6. A few molecular markers that are thought to be associated with artemisinin resistance have been suggested and identified (Uhleman et al., 2005; Krishna et al., 2004; Li et al., 2005; Wang et al., 2010). There is limited information on the field identification and application of these polymorphisms, as well as their relationship with clinical outcomes. However, nucleotide sequencing has shown extensive polymorphism in the gene, with no obvious relation to resistance (Menegon et al., 2008; Jambou et al., 2010). The fact that artemisinins also binds to ATPase6 orthologues in Toxoplasma gondii (Nagamune et al., 2007) and Trypanosoma cruzi (Mishina et al., 2007) necessitates continued molecular analysis of this candidate gene in malaria endemic areas. Studies on Co-artem done in various parts of Uganda since 2004 showed very good efficacy and effectiveness data confirming its usefulness as a first line treatment option (Yeka et al., 2008; Zurovac et al., 2008). Accordingly the UNMCP also recommended the inclusion of Co-Artem in the home based management of fevers, although there is concern about the overuse of the drug (Staedke et al., 2009). There is concern that if co-artem is used extensively, this may quickly lead to resistance and render this potent drug ineffective within a very short time yet there are no simple molecular tools that can be used to monitor the rate of development of resistance over the subsequent years of treatment policy implementation. Some other drugs, such as suramin, that bind to and inhibit ATPase’s are widely used and possibly increase the selection pressure for mutations in the proposed target gene (Emmick et al., 1994). Therefore, it is possible that the use of suramin has selected for PfATPase6 variants with diminished susceptibility to inhibitors, and such variants may be revealed by screening samples collected before introduction of Co-artem. The objective of this study was to develop a method to be used to determine in PfATPase6 and use it to determine the prevalence of mutations in PfATPase6 in P. falciparum parasites from Kasangati and Jinja in Uganda, at the time immediately preceding the policy change. Polymorphisms in amino acids 260-266, 769 and 1039 of PfATPase6 of P. falciparum parasites from Kasangati and Jinja, Uganda were studied. Materials and Methods Samples Three hundred stored blood spotted filter papers collected between the years 1999 and 2004 were analyzed for possible presence of mutations that are associated with co-artem resistance. The clinical information regarding these samples has been reported previously (Sendagire et al., 2005). These samples were collected to study resistance to CQ and SP but new approvals for subsequent molecular analysis of drug resistance were obtained from the ethical committee to allow further evaluation on resistance to Co-Artem. Twenty samples for 1999 were available, as were 59 samples for each of 2001 and 2004, the remaining 162 were for 2003. DNA extraction and PCR DNA was extracted using the chelex method (Plowe et al., 1995). Primers specifically designed to amplify 3 regions of the gene covering amino acids K260, L263, Q266, S769 and N1039 were used for PCR. Primers for PCR to amplify 2 segments of exon 1 and the whole of exon 2 were designed based on PfATPase6 sequence (AC number Q08853, AJ532679) using the Primer3 program (www.primer3.com). All primers were manufactured by MWG, Germany. The PCR strategy is shown in figure 1 and primer sequences (Table 1). Table 1: Nucleotide sequences of primers used for detection of mutations in Plasmodium falciparum ATPase6
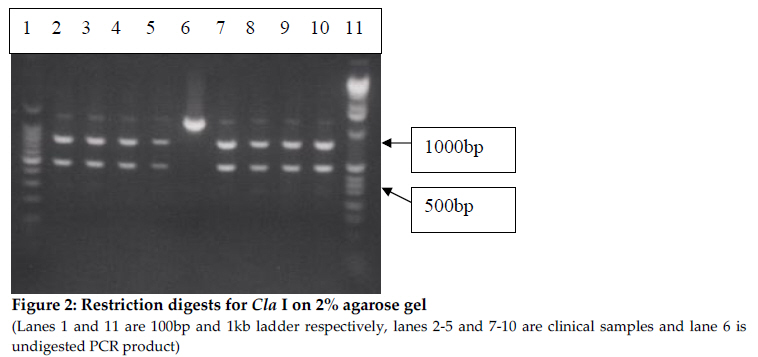
* Letters in bold represent changes from original sequence Conditions for outer PCR were: Initial 940C for 2 minutes, followed by 40 cycles (denaturation at 940C for 15 seconds, annealing at 500C for 30 seconds, strand extension at 720C for 2 minutes) and final extension at 720C for ten minutes. In a 50 µl PCR reaction, the extract of DNA (4µl) for outer and (2µl) for inner PCR was mixed with 2mM MgCl2 (Fermentas), 250mM dNTPs (Invitrogen), 1X PCR buffer (Fermentas), 150 nM of each primer and 1 unit of Taq Polymerase (Fermentas). PCR conditions for the inner PCR were similar but the annealing temperature was 450C and the final concentration of MgCl2 was 2.5mM. DNA from the laboratory strains 3D7 and HB3 (from MR4) were used as positive controls and sterilized distilled water was used as a negative control. All experiments were done in triplicates. Restriction fragment length polymorphisms (RFLP) The restriction map for PfATPase6 was generated using DNA for windows. PCR products were obtained and digested with the enzymes ClaI, HincII and Tsp509I. ClaI cuts the wild type sequence only once within the 1280 bp exon 1 PCR product resulting in 2 bands, and the recognition sequence covers codons I261 and D262 HincII cuts the PCR product four times (position 236, 551, 573 and 796) resulting into 5 segments. The recognition sequence for HincII covers the codons for G265 and Q266 and a mutation will lead to 4 bands of size 256, 315, 22 and 687bp. The PCR product for exon 2 is cut by the Tsp509I enzyme at nucleotide positions 56, 92, 193 and 291 and results into 4 bands. One of the cleavage sites is disrupted if there is a mutation at position 56-60 and we expect only 3 bands of size 92bp, 98bp and 101bp. All restriction enzymes were obtained from New England Biolabs. The buffers supplied with the enzymes were used. 10µl of the PCR product from segment 1 of exon 1 was digested with 1U of either ClaI or HincII at 37°overnight, followed by thermal inactivation by incubating at 65oC for 20 minutes. 10 µl of the exon 2 product was digested with 1 unit of Tsp509I at 65°C for four hours. Digests were run on 2%-2.5% agarose gel and visualized under UV transillumination after treatment with ethidium bromide. Polymorphisms at 260, 263 and 769 analysed by mismatching primers containing engineered restriction sites At codons 260, 263 and 769 there are no suitable overlapping cleavage sites. In order to detect possible polymorphisms at codons 260 and 263, specific restriction cleavage sites were introduced by modified primers as shown in Table 1. In both cases cleavage sites for MseI are generated if the sequence is wild type, and a mutation at either codon would destroy those cleavage sites. For position 769 primers have been designed by Sisowath et al to generate a cleavage site for RsaI (Sisowath et al., 2007) and these were used in this work. Construction of positive controls To be able to show that the methods designed really differentiates between mutant and wild type sequences site-directed mutagenesis were performed on cloned PCR products to generate plasmids containing mutant variants of the sequence. Products from both segments of exon 1 were cloned directly after PCR with the pJet Cloning Kit (Fermentas, Lithuania). The cloned PCR products were then subjected to site-directed mutagenesis with the Quick-Change protocol (Invitrogen). Primers are shown in table 1. PCR-conditions for the mutagenesis was: A 50µl reaction containing Pfu buffer, 200µM dNTP mixture, 0.5µM of each primer and 2.5 units of Pfu polymerase was processed in an Eppendorf Thermocycler with the following programme: Initial 95°C for 30 seconds followed by 17 cycles with denaturation at 95°C for 30 seconds, annealing at 55°C for 1minutes and strand extension at 68°C for 7 minutes. DNA sequencing DNA sequencing of 15 samples covering the 960bp of segment 2 of exon 1 was done using the BigDye Terminator labeled cycle sequencing kit (Applied Biosystems) and an ABI prism 310 Genetic Analyzer (Applied Biosystems). Results The primers designed were able to give good PCR products in 296 of 300 samples, which is more than 98% of all samples studied. In the first analysis, the PCR product covering segment 1 of exon 1 was digested with ClaI and HincII. The PCR product covering exon 2 was digested with Tsp509I. Restriction digestion with ClaI enzyme, at codons 261-262, HincII at codons 265-266 and Tsp509I at codon 1039 showed no mutation in the 296 samples tested (Figures 2, 3 and 4). In order to evaluate the methods used in the absence of any mutations in the samples analysed we produced control plasmids. The product from inner PCR of the first segment of exon 1 was cloned in the vector pJET and subjected to site-directed mutagenesis to generate plasmids with the respective nucleotide polymorphisms. The change in the resulting plasmids was confirmed by nucleotide sequence determination. The wild type and mutationally changed plasmids were then used with the primers generating additional restriction sites at the studied positions. The results show clearly that the introduction of additional cleavage sites gives good cleavage of the wild type but not of the mutationally changed plasmids (Figures 5 and 6). Figure 5 shows that PCR products from wild type and mutated plasmids are digested by MseI after amplification with the primer 260introfw, which introduced a changed nucleotide from A to T. The wild type plasmid produced four cleavage products, two of which (70 and 57 or 56) were visible. One of the cleavage sites of the plasmid with mutation at position 260 is mutated and MseI cuts at three positions instead of four, two of which (100 and 57 or 56) were visible and the 100 bp band is distinctly different from the 70 bp band. Digestion of the plasmid with mutation at position 263 was included as a control and was identical with the wild type plasmid as expected. Figure 6 shows that the PCR product from the wild type plasmid is digested by MseI when amplification was done with the primer 263introrev, which introduced a changed sequence from T to A. The PCR product from the wild type plasmid contains one single cleavage site and the larger fragment can be seen in the picture. The cleavage site in the plasmid mutated at position 263 (from TTAT to GGAT), is destroyed and undigested products are seen in the figure. When the clinical samples were analysed with the same procedure, all PCR products both for codon 260 and codon 263 were digested by MseI and thus no mutations were found. The nucleotide sequences of PCR products of the second segment from exon 1 in 15 randomly selected clinical samples revealed no mutation in these parasites from Kasangati and Jinja. For the rest of the samples we employed a RFLP analysis by the method of Sisowath et al., 2007). Also in this case, only wild type results were scored. No single sample with mutation at codon 769 was found. Discussion So far there has not been any solid evidence of artemisinin resistance (Xiao et al., 2004) although recent reports from Cambodia show slower parasite clearance during artemisinin treatment. This may indicate beginning tolerance to the drug. However, there is concern that the free provision of artemisinins in the government health units after the policy change may lead to appearance of artemisinin resistance if careful supervision of distribution and usage of these drugs are not taken care of. We have previously reported that there was a high rate of increase of mutations in DHPS and DHFR probably due to the indiscriminate use of SP. The main purpose of this study was to design simple molecular methods for screening of samples for the presence of mutations that may lead to artemisinin resistance. Since no definite link between particular mutations and resistance development, we focused on positions in the protein that have been implicated in artemisinin binding. The interaction between the amino acids K260 and N1039 is important in the gate keeping action of the artemisinin binding pocket of PfATPase6 (Uhleman et al., 2005). The L263 was early on indicated as a possible mutation spot because of differences between parasite and humans (Uhleman et al., 2005). Q266 is among the amino acids which are important in binding the artemisinins inside the binding pocket; therefore mutation at this site may impair the effective binding of these drugs and hence lead to drug resistance. These positions were chosen for the present study. However, no restriction enzyme cuts across codons 260 and 263, so here we designed an alternative method using introduced restriction site polymorphism as has already been done for position 769. In conjunction with this we also constructed positive control plasmids with engineered mutated sites to include in the analyses. The primers designed in this study were able to amplify the desired regions of the gene and generate good bands. In those cases where we had generated control plasmids, we could clearly see the difference between wild type and mutant. This study shows that there is no mutation at all the codons tested, i.e codons 260-263, 266, 769 and 1039. This is consistent with what was found in Cambodia, where there was no mutation in PfATP6 field isolates 1 year after policy change to ACT (Jambou et al., 2005). Also Afonso et al 2006 found no mutations in ATP6 of P. Chabaudi even after selection of parasites that could survive treatment. They later showed that the resistant parasites had changes in another gene coding for a deubiquitinating enzyme (Hunt et al., 2007). Lack of mutations in PfATP6 does not exactly mean that there is no drug resistance due to lack of enough studies that have correlated this. Mutations in a multidrug resistance gene Pfmdr1 in these parasites was not analysed and some studies have shown selection of mutants with 86Y by use of co-artem (Sisowath et al., 2005). The simple method for detection of mutations in PfATP6 developed in this study could be adopted for continued monitoring of parasites from the field, although it gives somewhat limited information, since there is no established drug resistance marker for artemisinin resistance and even reports about changes in pfATP6 are not conclusive. Thus to obtain enough information, full sequencing of the gene would be necessary. Sequencing of a few samples at these amplified regions did not show any of the mutations reported by other studies (Jambou et al., 2005). A continued search for more sites that are likely to mutate in PfATP6 using full sequencing is also important, especially in cases where treatment failure or recrudescence is seen. Meanwhile, the restriction enzyme digestion assays give good result and can be used for monitoring PfATP6 polymorphisms in a part of the protein that is important for interactions with artemisinin. References
Copyright 2011 - Tanzania Journal of Health Research The following images related to this document are available:Photo images[th11007f3.jpg] [th11007f4.jpg] [th11007f2.jpg] [th11007f5.jpg] [th11007f6.jpg] [th11007f1.jpg] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}