 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
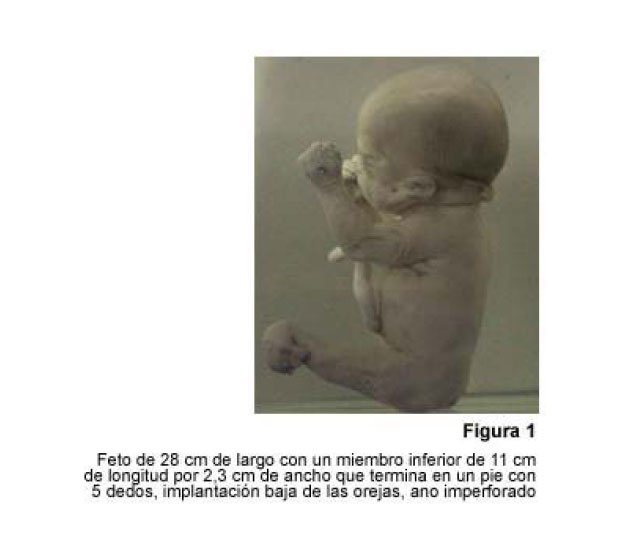
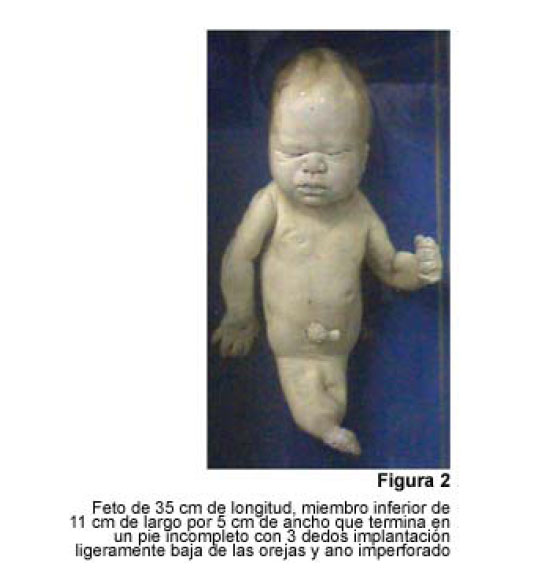
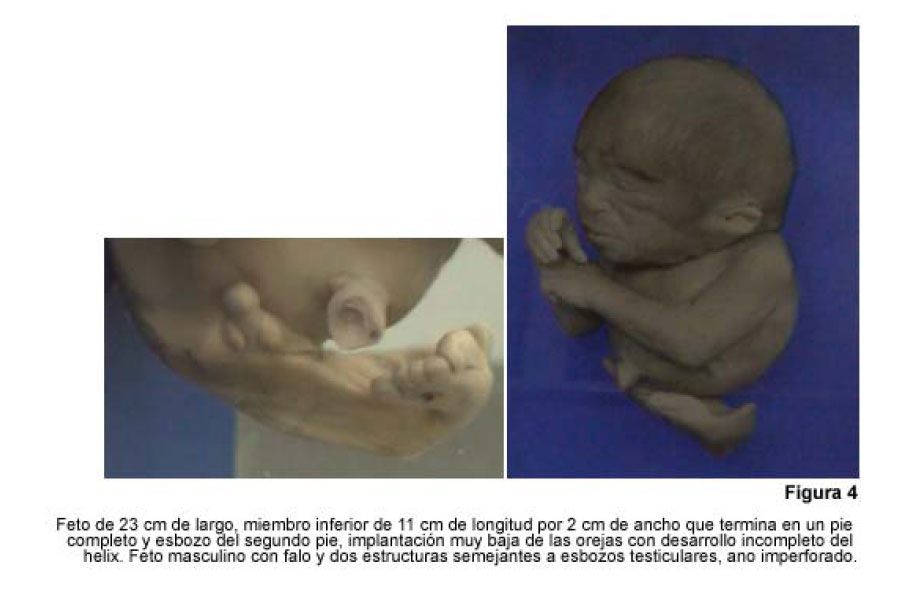
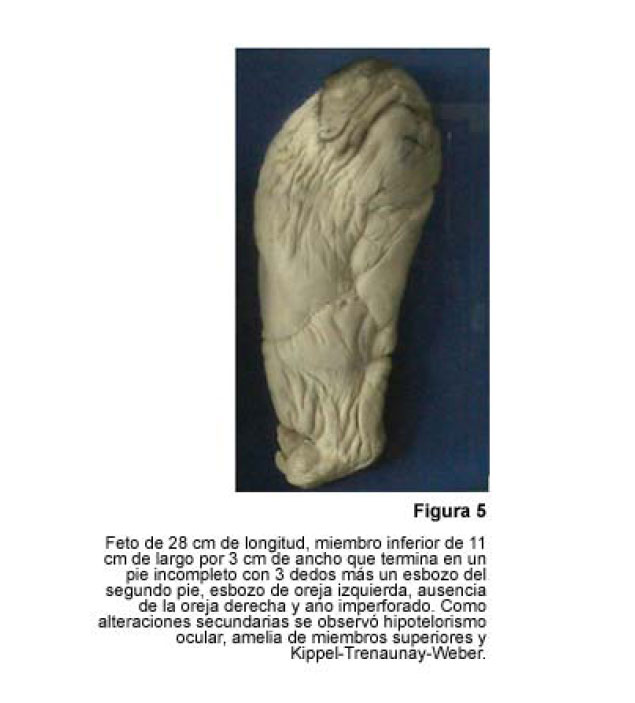
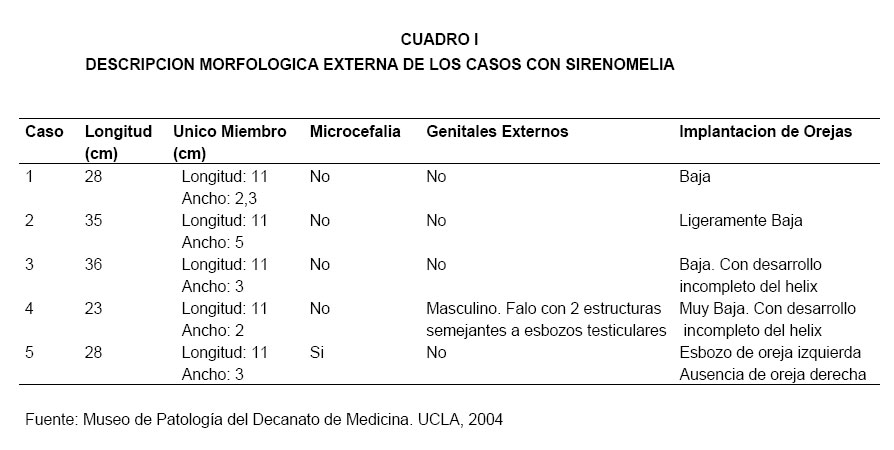
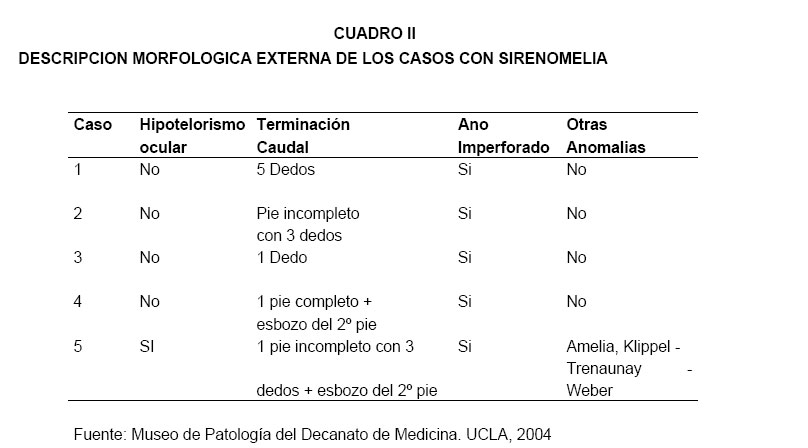
VITAE Academia Biomédica Digital, No. 24, Julio-Septiembre, 2005 Sirenomelia. Estudio de cinco casos y revisión de la literaturaSirenomelia. Report of five cases and a review of the literature.Autores: Bracho V, GA.*, Tovar J.R.*, Rodríguez M. **, Moreno B. *** *Anatomopatólogo. Profesor Titular. Decanato de Medicina.
U.C.L.A. Correo
Electrónico. gustavo_bracho@hotmail.com. Fecha de Recepción 20 Mayo - Fecha de Aceptación 22 Junio Code Number: va05012 RESUMEN: La Sirenomelia es una patología extremadamente rara; se produce por la fusión de los miembros inferiores secundaria a un trastorno severo en el desarrollo del blastema caudal axial posterior (en la cuarta semana del desarrollo embrionario), posiblemente debido a una alteración vascular de una de rama de la arteria aorta abdominal. Se presenta en forma aislada o asociada (a trastornos renales, cardiovasculares, gastrointestinales, respiratorios neurológicos o genitales), formando parte de Síndrome de Regresión Caudal. Se describen 5 casos de recién nacidos con Sirenomelia provenientes del Museo de Patología de la Facultad de Medicina de la Universidad Centroccidental “Lisandro Alvarado”, Barquisimeto, Venezuela. PALABRAS CLAVE: Sirenomelia, malformación congénita, desarrollo embrionario. SUMMARY: Sirenomelia is an extreme rare pathology. It is produced by inferior limbs fusion, secondary to a severe disorder in the posterior axial caudal blastema development (during the fourth week of embryonic development) possible owing to a vascular alteration of a branch of abdominal aorta artery. It presents as an isolated or associated form (renal, cardiovascular, gastrointestinal, respiratory, neurological or genital disorder) constituting part of the Caudal Regression Syndrome. We describe five cases or Sirenomelia newborn from the Pathology Museum at Faculty of Medicine, Universidad Centroccidental “Lisandro Alvarado”, Barquisimeto, Venezuela. Introducción La Sirenomelia es una malformación congénita letal extremadamente rara descrita desde mediados del siglo XIX. Se caracteriza por la fusión completa o parcial de los miembros inferiores, causada por un defecto del blastema caudal axial posterior en la cuarta semana del desarrollo embrionario (1). Puede presentarse de forma aislada, asociada con severas malformaciones urogenitales y gastrointestinales o formando parte del Síndrome de Regresión Caudal (2). La incidencia es de 1 de 60.000 nacimientos vivos (3), con una relación masculino-femenino 3:1 (4), y el cariotipo es normal: 46XX o 46XY (5). Existen varios sinónimos de esta malformación: sirenomelus, monopodia, sirena, simelia, uromelia, feto sireniforme, feto cuspídeo, simpodia, sympus, síndrome de Mermaid o de la sirena, síndrome de Vater, anomalía de Duhamel. (6) La etiología precisa de la Sirenomelia no es bien conocida y aunque muchas teorías han sido propuestas ninguna se ha considerado conclusiva. (7, 8) Teoría de la falla primaria que sugiere la presencia de un defecto primario en el desarrollo de las somitas caudales, determinando la no inducción de un número de ellos, que originan deficiencias en la porción distal del embrión. Teoría del déficit nutricional que sostiene que existe un compromiso de la perfusión sanguínea en la región caudal del cuerpo, por la obstrucción o anomalía del sistema vascular arterial correspondiente. Finalmente la Teoría mecánica que plantea que el desarrollo caudal anómalo es debido a una fuerza intrauterina en el extremo caudal del embrión. Stevenson et al (7) propuso la teoría de la presencia de una arteria vitelina persistente consecuencia de una alteración en el desarrollo vascular temprano durante el día 22-23 del desarrollo embrionario, así en vez de que la sangre regrese a la placenta a través de las arterias umbilicales pares que se derivan de las arterias ilíacas, la sangre retorna a la placenta, se produce una falta de perfusión y por lo tanto las somitas inferiores no se desarrollan normalmente, lo explica algunas de las anormalidades presentes en los fetos Sirenomélicos (9, 10). No se han determinado factores genéticos causales (11) , sin embargo se ha relacionado con la exposición a agentes teratógenos (3) como la vitamina A (a dosis excesivas antes de la 4ta semana del desarrollo) (12), la exposición a cocaína durante la mayor parte del primer trimestre del embarazo (13) y la hiperglicemia (14, 15, 16, 17, 18) en madres diabéticas, aunque la literatura actual refiere que solo de un 0,5-3,7% de los casos de Sirenomelia ocurren descendientes de madres diabéticas (18, 19, 20). El objetivo de este trabajo es la de hacer un aporte al estudio de la Sirenomelia como parte del Síndrome de Regresión Caudal. Descripción de Casos Los casos descritos corresponden a cinco casos de Sirenomelia pertenecientes al museo de Patología del Decanato de Medicina de la Universidad Centroccidental Lisandro Alvarado. No se reportan lesiones internas dados que son casos de larga data y no se dispone de historia clínica. La descripción es externa (Ver cuadro I y II). Caso 1: Feto de 28 cm de largo con un miembro inferior de 11 cm de longitud por 2,3 cm de ancho que termina en un pie con 5 dedos, implantación baja de las orejas, ano imperforado. (Foto Nº 1) Caso 2: Feto de 35 cm de longitud, miembro inferior de 11 cm de largo por 5 cm de ancho que termina en un pie incompleto con 3 dedos, implantación ligeramente baja de las orejas y ano imperforado. (Foto Nº 2) Caso 3: Feto de 36 cm de largo, miembro inferior de 11 cm de longitud por 3 cm de ancho que termina en forma aguzada con 1 solo dedo, implantación baja de las orejas con desarrollo incompleto del helix y ano imperforado. (Foto Nº 3 ) Caso 4: Feto de 23 cm de largo, miembro inferior de 11 cm de longitud por 2 cm de ancho que termina en un pie completo y esbozo del segundo pie, implantación muy baja de las orejas con desarrollo incompleto del helix. Feto masculino con falo y dos estructuras semejantes a esbozos testiculares, ano imperforado. (Foto Nº 4 ) Caso 5: Feto de 28 cm de longitud, miembro inferior de 11 cm de largo por 3 cm de ancho que termina en un pie incompleto con 3 dedos más un esbozo del segundo pie, esbozo de oreja izquierda, ausencia de la oreja derecha y ano imperforado. Como alteraciones secundarias se observó hipotelorismo ocular, amelia de miembros superiores y Klippel-Trenaunay-Weber.(Foto Nº 5 ) Resultados Los resultados están descritos en los Cuadro I y II que se anexan y se refieren a los casos señalados en las Figuras 1 a 5. Discusión Sympodia es el término dado por Ballantyne a una malformación congénita caracterizada por un “estado de inversión de las extremidades inferiores con una fusión mayor o menor de las partes, y con un desarrollo imperfecto de ellas y los órganos pélvicos vecinos así como la pelvis” (21) El Síndrome de Regresión Caudal (SRC) propuesto por Duhamel en 1961 consiste en un defecto primario que ocurre en el mesodermo del eje medio posterior del embrión y permite la fusión de los primordios de los miembros en sus márgenes fibulares, con ausencia o desarrollo incompleto de las estructuras caudales intercurrentes (22), y que se extiende a varios niveles craneocaudales(23); como consecuencia se presenta un espectro de malformaciones congénitas donde hay la alteraciones esqueléticas (agenesia lumbosacra) combinada con deformidades variables de los miembros inferiores y malformaciones del tracto digestivo, genitourinario (14, 24) y deterioro neurológico (25, 26). Se han sugerido como causas: la diabetes materna, la hipoperfusión vascular y la predisposición genética (22). La clasificación de la Sirenomelia separada del SRC es todavía discutida, y aunque los avances en el entendimiento del patrón mesodérmico axial durante el desarrollo embrionario temprano sugiere que la Sirenomelia representa la forma más severa terminal del SRC (5, 22, 27, 28, 29), existen hallazgos actuales confirmados de recientes estudios en patología pediátrica que sugieren que estas dos condiciones son entidades separadas (14, 30). La Sirenomelia se clasifica en : 1.- Simelia Apus: fémur y tibia únicos con ausencia de pies (1 tibia y 1 fémur, sin pies) 2.- Simelia Unipus: fémures, tibias y peronés normales con fusión parcial de los pies (2 tibias, 2 fémur, 2 peroné, 1 pie) 3.-Simelia Dipus: ambos pies presentes con apariencia de aletas (2 piernas fusionadas, 2 pies). (Foster, 1865) (7, 31) En la actualidad se ha informado de una clasificación más detallada, agrupando las variedades en 7 subtipos: (32, 33) Tipo I: pares de fémur, tibia y peroné presentes. Tipo II: peroné único fusionado. Tipo III: ausencia de peroné. Tipo IV: fémures parcialmente fusionados con peroné único. Tipo V: fémures parcialmente fusionados con peroné ausente. Tipo VI: fémur y tibia únicos. Tipo VII: fémur único con ausencia de tibia y peroné. En el presente trabajo solo se reportan lesiones externas, así el caso 1 presenta un miembro inferior único que termina en un pie con 5 dedos, el caso 2 termina en un pie incompleto con 3 dedos, el caso 3 termina en forma aguzada con 1 solo dedo, en el caso 4 termina en un pie completo y esbozo del segundo y en el caso 5 pie termina en un pie incompleto con 3 dedos más un esbozo del segundo pie. Llama la atención la medida única del miembro inferior de 11 cm de longitud en todos los casos, sugiriendose que la malformación ósea no este directamente asociada a la patología visceral descrita. Las anomalías asociadas a Sirenomelia son: la presencia de una arteria umbilical única (usualmente la derecha) en continuación directa con la aorta abdominal (34), el sistema gastrointestinal es anormal en todos los casos (1/3 de los casos tienen anomalías del tracto gastrointestinal alto, incluyendo divertículo de Meckel, agenesia de la vesícula biliar y atresia duodenal; y en todos los casos hay agenesia del colon terminal y ano imperforado). Todos los casos presentan ano imperforado. Trastornos del sistema genitourinario (2/3 de los casos tienen agenesia renal bilateral y el resto displasia renal quística (4, 35), los uréteres y la vejiga urinaria por lo general están ausentes y cuando se presentan son hipoplásicos). Análisis de la literatura y estudios comparativos señalan la relación entre el grado de fusión de los miembros inferiores rudimentarios y la intensidad del subdesarrollo de los órganos urogenitales cuando los ductos mesonéfrico y paramesonefrico están involucrados (36). Según Barr (37), si el ducto mesonéfrico progresa lo suficiente para alcanzar los brotes ureterales y penetrar en el blastema metanéfrico, los riñones se pueden formar. Cuando la porción metanéfrica del mesodermo intermediario es defectuosa, se pueden presentar riñones anormales o hipoplásicos. Defectos cardíacos (1/4 de los infantes tienen defectos ventriculoseptales), a nivel del tracto respiratorio (hipoplasia pulmonar, casi constante debido al oligohidramnios; los pulmones pueden ser lobulados) (38), ambigüedad o ausencia de genitales externos, aunque las gónadas, por lo general bilaterales, pueden ser identificadas en el 80% de los casos. El caso 4 es un feto masculino con falo y dos estructuras semejantes a esbozos de testículos. Tórax malformado, defectos de las vértebras sacras inferiores, alteraciones del tubo neural (mielomeningocele, malformación de Arnold-Chiari, hidrocefalia) (39) , anomalías de la pared abdominal, oligohidramnios o anhidramnios severo, facies de Potter (38, 40), manifestaciones extrarrenales del Síndrome de Potter (41). Se han descrito casos de fístula traqueoesofágica y hernia diafragmática (38). La asociación de VATER es la combinación de defectos morfológicos incluyendo defectos vertebrales, atresia anal, fístula traqueoesofágica, atresia del esófago, anomalías renales y radiales, y se sugiere que para ser catalogado como Síndrome de VATER debe presentar por lo menos 3 de estos defectos (1). La literatura señala la asociación entre Sirenomelia la secuencia VATER (20, 24, 29, 33). La asociación de VACTERS es una poco común forma expansiva de la asociación de VATER que incluye defectos cardíacos, de los miembros e hidrocefalia . Un estudio realizado de la base de datos de 1 millón de nacimientos con malformaciones congénitas investigados en 11 países de América Latina concluye la Sirenomelia y VACTERS son condiciones con patogénesis similar (1, 42). Además, la presencia de tejido heterotópico renal en la pared del colon soporta el hecho de que la asociación de VACTERS y la Sirenomelia representan entidades relacionadas. (43) Cerca del 20 % de los casos descritos son producto de embarazos gemelares (19 ,20 ,44, 45). La incidencia es 100-150 veces mayor en gemelos monocigotos comparada con gemelos dicigotos (40, 46, 47). Ocasionalmente se han señalado raras asociaciones con fetos sirenomelicos: glándulas adrenales ectópicas (aberrantes), de tamaño y posición normal, sin desarrollo del sistema urinario (48). Situs inversus completo. (38, 49) .Pentalogía de Cantrell en gemelos monocigotos (50) .Raquisquisis y Craneoraquisquisis total (se han reportado solo 6 casos) (51) . Síndrome pared corporal extremidades (52). Ciclopía (53). Espina bífida (54). Hernia umbilical (55, 56) .Sirenomelia con tetrasomía 13 (57) . Ventriculomegalia secundaria a malformación Chiari II (58). Secuencia de disrupción de cordón o banda amniotica (59). Anencefalia (sólo 3 casos reportados hasta 1999) (16, 60) .Cebocefalia y holoprosencefalia alobar (61) .Síndrome de la apertura de la pelvis inferior (62). Paladar hendido, duplicación de genitales internos(60). Onfalocele, cloaca persistente y 2 riñones normales (63) . El caso 5 tiene amelia de miembros superiores, hipotelorismo ocular y Klippel-Trenaunay-Weber. Además en todos los casos hay implantación baja de las orejas. Los casos reportados en la literatura médica han sido siempre asociados a muerte perinatal, la sobrevivencia es extremadamente rara y solo es posible en la ausencia de agenesia renal bilateral (cuando los riñones están formados y no son severamente displasicos) ( 41, 64). Las referencias encontradas de sobrevivientes son: Infante de 4 años de edad, con ausencia de vejiga urinaria, aorta distal anormal (9). Niña de 4 años con unidades renales funcionales (65). Infante de 3 meses de edad con displasia renal. (66) Neonato a término con riñones pélvicos bilaterales fusionados con displasia renal, displasia pelvica y sacra, anormalidades genitales (67) . La Sirenomelia debe ser sospechada en el periodo prenatal en caso de presentarse con oligohidramnios severo y retardo del crecimiento intrauterino (3). Para realizar el diagnóstico prenatal de la Sirenomelia se utiliza las radiografías simples de abdomen, la resonancia magnética (68) y la ultrasonografía transvaginal. El diagnóstico con ultrasonografia transvaginal se puede realizar desde la semana 13 de gestación (22,64, 69, 70) . En el tercer trimestre del embarazo el diagnóstico resulta difícil debido a la presencia de oligohidramnios severo relacionado con la agenesia renal bilateral, sin embargo durante el 2º trimestre la cantidad de líquido amniótico debería ser suficiente para permitir el diagnóstico (27, 32, 71, 72) . Sirtori, en una revisión de 11 casos comprobados demostró una incidencia de oligohidramnios de 45% (73) por lo que la amnioinfusión o infusión del líquido amniótico artificial es de ayuda cuando existe oligohidramnios severo (57, 68, 74) El diagnóstico por ultrasonido puede demostrar la presencia de una sola extremidad (visualización de ambas tibias y peronés, con o sin pies, o la identificación de un fémur único pueden servir de clave), oligohidramnios y agenesia renal bilateral (6) y aunque el diagnóstico es entorpecido por la extrema flexión fetal y la pobre visibilidad, eventualmente se pueden observar defectos cardíacos(4 casos), de la pared abdominal ( 4 casos) y defectos esqueléticos (10 casos) (73) .La simelia tipo dipus le agrega dificultad al diagnóstico ya que los huesos de los muslos y de las piernas están totalmente formados, de hecho estudios sugieren que el diagnóstico solo se puede hacer cuando hay tipos apus o unipus de simelia (49) La radiografía en la autopsia permite la categorización en base a la deformidad del esqueleto (27). Para la determinación del sexo en fetos Sirenomelicos con genitales ambiguos se uso PCR (Reacción de la cadena de polimerasa) directa contra el gen SRY. El PCR basado en la determinación del sexo es una rápida técnica en pacientes con estas anomalías. (75) Referencias
Copyright 2005 - Centro de Análisis de Imágenes Biomédicas Computarizadas CAIBCO, Instituto de Medicina Tropical – Facultad de Medicina, Universidad Central de Venezuela The following images related to this document are available:Photo images[va05012t1.jpg] [va05012f4.jpg] [va05012f1.jpg] [va05012t2.jpg] [va05012f5.jpg] [va05012f3.jpg] [va05012f2.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}