 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
VITAE Academia Biomédica Digital, Número 32, Julio-Septiembre, 2007 Microbiología Helicobacter pylori: Un Camino Al Cáncer Helicobacter pylori: a way to cancer Sabrina Anselmi1 , Víctor Castillo2 , Katiuska González3 , Angel Guillermo4 , Sathyn La Rosa5 , Yugel Medina6 , Marcel J. Marcano-Lozada7
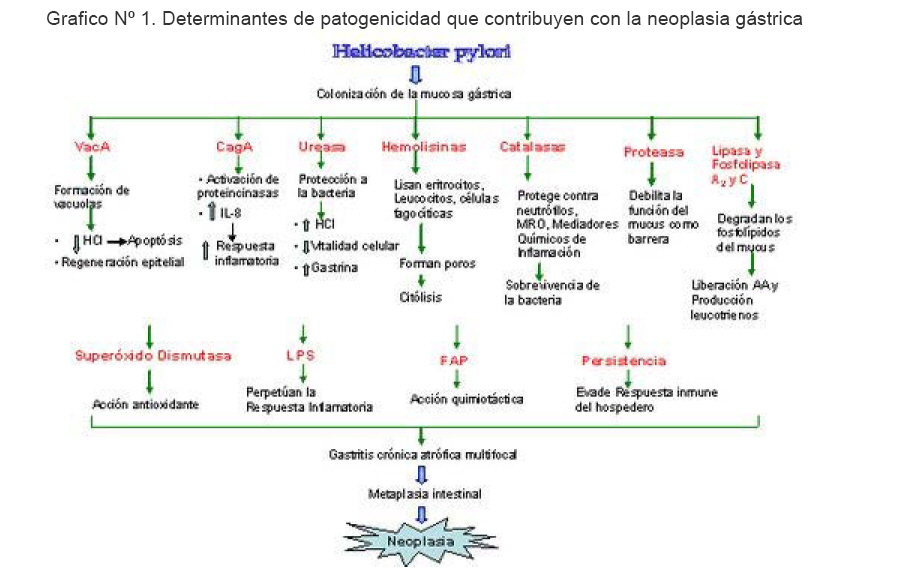
1 Escuela de Medicina “José María Vargas”, Facultad de Medicina, Universidad Central de Venezuela. Fecha de recepción: 19/11/2008 Fecha de aceptación: 10/02/2008 Code Number: va07026 Hace más de dos décadas se estableció la asociación entre infección por Helicobacter pylori y la aparición de gastritis y úlcera péptica. Actualmente, se ha encontrado un nexo entre H. pylori y procesos oncogénicos como el carcinoma gástrico o linfomas MALT. Este nexo adquiere cada vez más importancia y complejidad, ya que se han encontrado factores intrínsecos de la bacteria que influyen en el desarrollo de la oncogénesis tales como la presencia de CagA, VacA, ureasa, BabA2, hemolisinas, b-catenina, gastrina, mucinasa, catalasa, proteínas del shock térmico, lipasa, y factor activador de plaquetas. Los mecanismos por los cuales el microorganismo causa un proceso patológico que evoluciona hasta el carcinoma gástrico también dependen de aspectos medioambientales y propios del hospedero (antecedentes genéticos, respuesta inmune, reinfección, dieta, ingesta de alcohol y uso de tabaco). La infección gástrica por H. pylori es muy común, encontrándose una prevalencia de un 80-90% de la población, por lo cual muchos pacientes desarrollan gastritis aguda, que puede evolucionar tras un proceso prolongado a gastritis crónica y úlceras pépticas, conllevando a la formación de zonas de metaplasia gástrica, que condicionan, por último, mayor riesgo de desarrollar cáncer gástrico. En el presente trabajo se revisan la influencia y asociación de la infección por H. pylori basados en los procesos patogénicos y factores de riesgo para el desarrollo de cáncer gástrico. Palabras Claves:H. pylori, apoptosis, adenocarcinoma, linfoma MALT, oncogénesis. Abstract More than two decades ago an association was established between the infection by Helicobacter pylori and gastritis and peptic ulcer. Presently, it has been demonstrated that this association involves much more than these pathologies, finding a nexus between the infection by H. pylori and oncogenic processes like gastric carcinoma or MALT lymphomas. It has been found that some intrinsic factors of the bacterium can promote the development of oncogenesis, such as the presence of CagA, VacA, urease, BabA2, hemolysins, b-catenine, gastrine, mucinase, catalase, heat shock protein, lipase, and platelets activator factor. The mechanisms by which this microorganism causes a pathological process that can evolve to gastric carcinoma, also depends of environmental aspects and host´s factors such as the genetic load, immune response, reinfection, diet, smoking habits and drinking alcoholic beverages. The infection by H. pylori in the gastric cavity is very common, with a prevalence of a 80-90% of the population, thus exists a great population susceptible to suffer pathologies caused by this infection, such as acute gastritis, that can evolve after a prolonged process to chronic gastritis and peptic ulcers that might be the beginning of zones of gastric metaplasia, that finally imply, a greater risk of developing gastric cancer. We review the influence and association of the infection by H. pylori in the gastric cancer; as well as host’s conditions that can influence that this infection evolves until triggering an oncogenic process. Key Word: H. pylori, apoptosis, adenocarcinoma, MALT lymphoma, oncogenesis. Introducción La prevalencia mundial de la infección por Helicobacter pylori (H. pylori) es aproximadamente del 80 al 90% en la población, donde cerca del 3% de los individuos presentará, en algún momento de su vida sintomatología atribuibles a dicha infección, tales como gastritis, úlcera péptica, cáncer o linfoma gástrico. (1) En este orden de ideas, cada vez se fortalece más el nexo entre el cáncer gástrico y la infección por H. pylori, llegándose a plantear la erradicación indiscriminada o la vacunación de la población en zonas de alto riesgo de cáncer gástrico(2). Aunque no se ha podido demostrar que esta bacteria produzca sustancias carcinógenas lesivas directamente sobre la mucosa del estómago, se sabe que estarían implicados diversos mecanismos indirectos que actuarían sobre la mucosa gástrica durante la infección (3). Esto ha estimulado la búsqueda de los cofactores para la progresión de la infección de H. pylori a la oncogénesis gástrica. En junio de 1994, la Agencia Internacional para la Investigación sobre el Cáncer, perteneciente a la Organización Mundial de la Salud, declara que la bacteria H. pylori es un carcinógeno de primera clase, lo que lo sitúa entre los agentes tumorigénicos más peligrosos (4). Entre los principales determinantes de patogenicidad de H. pylori pueden mencionarse: Urea, flagelos, adhesinas, catalasa, proteínas del shock térmico, mucinasas y Fosfolipasas, citotoxina A (CagA), toxina vacuolizante (VacA), FldA, BabA2. Los cuales serán detallados posteriormente (5), (6). Según lo indicado por datos epidemiológicos, la incidencia del cáncer gástrico varía según el área geográfica en relación con exposición a factores medioambientales, tales como tabaco, alcoholismo, factores genéticos, dieta, entre otros. Todos estos factores, aunados a que el cáncer gástrico ocupa la segunda causa más frecuente de neoplasias y en Venezuela es la primera causa de muerte por cáncer en varones, y la tercera causa de muerte por cáncer en hembras después del cáncer de cuello uterino y de mama, hablan de la importancia de este estudio para la prevención y tratamiento de infecciones por H. pylori . Desarrollo H. pylori ha sido identificada como la causa de gastritis aguda y crónica activa, enfermedad ulceropéptica y gastritis atrófica. Igualmente se correlaciona en la génesis del adenocarcinoma gástrico y linfoma MALT. (7) Entre los principales factores de virulencia los más estudiados son CagA y VacA. Existen evidencias de que las citotoxinas y enzimas codificadas por estos genes son responsables del daño directo a la mucosa epitelial. La presencia de cepas H. pylori CagA (+) y la respuesta serológica a la proteína CagA están asociadas al desarrollo de úlcera péptica y cáncer gástrico. La citotoxina codificada por el gen VacA, induce la formación de vacuolas. Esta citotoxina tiene la capacidad de inducir la apoptosis y la subsecuente regeneración epitelial con cambios celulares. La evidencia muestra daño directo a la mucosa epitelial mediada por citotoxinas y por enzimas codificadas por estos genes. (8) H. pylori es una bacteria que se adquiere usualmente durante la infancia. Su presencia en la mucosa gástrica genera una respuesta inmunológica celular y humoral, la cual es incapaz de eliminar la bacteria. (9) Luego de varios años de discusión, se acepta que H. pylori es el agente bacteriano responsable de la gastritis y las úlceras pépticas. No obstante se discute sobre el potencial patogénico de esta bacteria en relación con el cáncer gástrico. Tras diversos estudios se encontró un riesgo mayor de padecer cáncer gástrico en los casos serológicamente positivos para esta bacteria. El estímulo antigénico mantenido por largo tiempo en la mucosa, además de una serie de factores externos como serían una baja ingesta de vegetales ricos en antioxidantes naturales y embutidos ricos en nitritos, todo ello aunado a factores de virulencia de H. pylori , podrían desencadenar una serie de eventos en la mucosa gástrica que pueden evolucionar de una gastritis superficial a un adenocarcinoma. (10) Existen estudios donde se describe que el reflujo biliar y H. pylori tienen un papel sinérgico en la proliferación celular. Se ha sugerido que el pH alcalino intragástrico inducido por el reflujo biliar produce un microambiente desfavorable para la colonización por H. pylori . Por ello algunos autores piensan que esta patología puede actuar como “protección” contra la infección bacteriana (11) Con respecto a las dispepsias no ulcerativas, se ha encontrado en diversos estudios, H. pylori asociado con un 43 a 87%, donde un porcentaje importante de los síntomas desaparecen o se reducen con la erradicación de la bacteria. (12) H. pylori coloniza e infecta la mucosa gástrica y constituye el agente causal más comúnmente asociado a dicho cáncer. Esta bacteria provoca gastritis crónica atrófica multifocal con hipoclorhidria, disminución del ácido ascórbico, lo cual facilita el sobrecrecimiento bacteriano y el aumento de nitrosaminas y nitrosamidas, que tienen alta capacidad mutagénica. La infección por H. pylori en la región antra se caracteriza por un infiltrado de neutrófilos marcado y mantenido que conduce a una atrofia focal, seguida por una metaplasia intestinal, y finalmente se presentan alteraciones epiteliales de tipo neoplásico. (12) Existe un incremento en la incidencia de aislamientos de H. pylori que muestran resistencia a varios de los antibióticos utilizados en las terapias de erradicación incluyendo claritromicina y metronidazol. H. pylori parece ser un factor más en la cadena causa de cáncer gástrico e incluso ha sido considerado como un carcinógeno definitivo (4). La respuesta inmune ante la infección por H. pylori es particularmente diversa, aunque no logra eliminar la bacteria ni evitar la reinfección luego de una exitosa terapia con antibióticos. Una vez establecida la infección, los antígenos bacterianos logran permear hacia los órganos linfoides periféricos y a nivel de mucosa, generando una respuesta de anticuerpos séricos IgG e IgA cuyas células secretoras se localizan junto con los linfocitos B. Adicionalmente, la presencia de esta bacteria provoca la progresiva acumulación de linfocitos T en el tejido gástrico proximal y distal. (13) La oncogénesis gástrica asociada a H. pylori se fundamenta en las acciones de sus múltiples determinantes de patogenicidad y la interacción que se produce entre la infección en el transcurrir del tiempo asociada a la respuesta inmunológica del hospedador. Determinantes de Patogenicidad de H. pylori que Influyen en el desarrollo de oncogénesis I CagA La proteína CagA es codificada por el gen PAI 30+, llamado así porque el contenido de guanina (G) + citosina (C) del PAI difiere del resto del genoma, y es un indicador de la presencia de la Isla de Patogenicidad Cag. Esta proteína es secretada dentro de las células del epitelio gástrico, igual que la citotoxina vacuolizante (VacA) por un sistema de secreción por contacto (tipo IV) también codificado en dicho gen. Dentro de las células epiteliales es capaz de inducir en la fosforilación de tirosinas en las proteínas celulares, induciendo una reorganización dinámica de la actina del citoesqueleto celular y la activación de múltiples proteínas que pueden alterar la transducción y transcripción de genes a nivel celular. (14) La infección por cepas de H. pylori CagA+ inducen la activación de cinasas dependientes de señales extracelulares; p38 y proteincinasas de actividad mitógena (MAP), así como también activa la transcripción de proto-oncogenes c-fos y c-jun. Debido a que las proteincinasas MAP regulan la proliferación y diferenciación celular, la muerte celular programada, el estrés celular y la respuesta inflamatoria, la activación de estas proteincinasas por la infección de H. pylori puede ser un factor esencial en la inducción de la inflamación y el carcinoma. El Factor nuclear -kB (NF-kB) es un factor regulador de la transcripción del gen para la producción de IL-8, esta demostrado que se activa frente a la infección por H. pylori, por la translocación nuclear de los heterodímeros p50/p65 y los homodímeros p50 de dicho factor. Este efecto producido tiene como consecuencia un incremento del IL-8mRNA, lo que traduce a un aumento en la producción de la IL-8. Se ha comprobado que las cepas de H. pylori CagA+ aumentan notablemente la expresión de IL-8 lo que induce por tanto una respuesta inflamatoria mayor. (15) Igualmente, causa un aumento inicial en la expresión de proteínas p53 y p21, seguido de un descenso, teniendo en cuenta que el gen p53 pertenece a la serie de genes de supresión tumoral, existe aquí una evidencia clara de su participación en procesos carcinogénicos, además, la expresión de Bcl-2 está aumentada, traduciéndose esto en una disminución de la apoptosis y un incremento persistente de la proliferación celular. Vac A El gen VacA está presente en todas las cepas de H. pylori, sin embargo, algunas de las cepas difieren considerablemente en la producción de la citotoxina vacuolizante. Esta variación está principalmente atribuida a la variación de la estructura del gen VacA. Las regiones de variabilidad se encuentran en el extremo 5`del gen (alelos s1 y s2) y en la región media del mismo (alelos m1 y m2) (16) Además, VacA codifica una proteína VIP-54 que induce una inhibición selectiva del CMH Clase II, comprometiendo así la presentación antigénica. (17) Por otro lado, disminuye la producción de ácido clorhídrico en el estómago, causando apoptosis en las células parietales y ayudando así en la infección. (4) En el estudio de la genotipificación de H. pylori se encuentra que la presencia del alelo s1 del gen VacA se correlaciona positivamente con la intensidad del infiltrado de polimorfonucleares. (1) Debido a está capacidad de promover la inflamación, las cepas con alelos s1/m1 están asociados a un mayor incremento en el riesgo de cáncer gástrico comparado con las cepas s2/m2. (16) Ureasa Una de las características más importantes de las cepas de H. pylori oxidasa y catalasa positivas, es la presencia de una potente enzima denominada ureasa, la cual protege a la bacteria de los efectos letales del ácido gástrico mediante la formación de una “nube de amonio” que le sirve para tamponar su entorno vital y poder colonizar el epitelio. La ureasa se localiza en el espacio periplasmático y en la membrana más externa de la bacteria; su actividad se beneficia cuando el pH es bajo. La producción de ureasa interviene en la regulación del metabolismo de la urea, forma dióxido de carbono y amoníaco. En diversos trabajos se señala la función tóxica del amoníaco sobre las células eucariotas de la mucosa gástrica, aunque algunos autores opinan que el amoníaco en sí no daña la célula sino que el daño es provocado por uno de sus metabolitos (denominado monocloramina), formado por la interacción del amoníaco con el ácido hipocloroso producido por los neutrófilos activados. El amoníaco producido por la ureasa difunde más fácilmente que el ion amonio, derivado de la unión del amoníaco y el ácido clorhídrico del jugo gástrico, por lo cual el amonio no se considera dañino para la mucosa gástrica. (17) En estudios experimentales, tanto con animales como en el hombre, infectados por H. pylori, la concentración de amoníaco en el jugo gástrico se encuentra elevada en comparación con los no infectados. Algunos autores reportan que el amoníaco causa daño mucosal porque, al actuar como agente necrotizante, altera el funcionamiento mitocondrial, la respiración celular y el metabolismo energético, con lo cual disminuye la vitalidad de las células y se produce su muerte. (17) El amoníaco es capaz de modificar la secreción gástrica al estimular la secreción de gastrina e incrementar la producción de ácido clorhídrico que alteran la barrera mucosa gástrica y con lo cual se favorece la retrodifusión de hidrogeniones y se provoca más daño hístico. Otros mecanismos que se involucran en el daño producido por el amoníaco sobre el epitelio gástrico son: inhibición de la liberación del factor estimulador de crecimiento epidérmico, potente efecto inhibidor del ciclo de Krebs, caída significativa de la mucina intercelular y alteraciones de la microcirculación gástrica (estasis), así como disrupción y necrosis de la capa superficial del epitelio. El amoníaco y otras sustancias liberadas por las bacterias son capaces de reducir la actividad bactericida de las células polimorfonucleares y de los monocitos, al inhibir la acidificación de los lisosomas durante la fagocitosis. (18) Determinantes de Patogenicidad de H. pylori que Influyen en el desarrollo de oncogénesis II Adhesina de unión a grupos sanguíneos (BabA2) Dos pasos son identificados en la patogénesis de H. pylori durante la colonización de la mucosa gástrica; éstos son la adhesión a la célula epitelial y la inducción de liberación de citocinas proinflamatorias (18). La adhesión de H. pylori a las células epiteliales gástricas es mediada por las adhesinas, entre ellas BabA que se une al epítopo B de Lewis del hospedero; ha sido postulado que la unión de BabA2 a las células gástricas puede facilitar la secreción de ciertos productos como la proteínas CagA hacia el epitelio y que está involucrada en el desarrollo de adenocarcinoma gástrico, úlcera peptica y gastritis severa (19). Las cepas BabA2 positivas contribuyen de forma directa en el proceso inflamatorio y de ésta manera es capaz de inducir un proceso neoplásico al contribuir en la secreción de citocinas proinflamatorias como IL-8 y aumentar la quimiotaxis de neutrófilos y de otras células linfocitarias. En estudios realizados, 80% de la población están infectados con cepas de H. pylori BabA2 positivos, lo cual se asocia con una inflamación crónica más intensa, presencia de atrofia y metaplasia intestinal del antro gástrico (20). Hemolisinas Son toxinas bacterianas que lisan eritrocitos a través de la disrupción de la pared celular y también son denominadas citolisinas (21). Las hemolisinas pueden lisar las membranas de otros tipos celulares, como por ejemplo, leucocitos del epitelio gástrico o células fagocíticas, lo cual produce daño tisular al haber liberación de peróxidos (22). H. pylori posee hemolisinas con diferentes funciones, lo cual depende de la composición de la membrana celular, según esto, las citolisinas, pueden clasificarse en tres categorías: enzimáticas (incluyendo las fosfolipasas PldA), formadoras de poro y surfactantes. Las citolisinas formadoras de poros, tales como TlyA, se unen primero a la membrana celular, una vez adheridas, la citolisina penetra y rompe la membrana a través de la formación de poros, lo cual conduce a alteraciones en la permeabilidad de la membrana y provoca finalmente la citólisis (23). Las hemolisinas de H. pylori contribuyen en la patogénesis de la infección. Algunas proteínas con actividad hemolítica como TlyA o PldA, pueden liberar mediadores de la inflamación y provocar agregación leucocitaria al lugar de infección y además pueden contribuir en la adhesión celular (22). Se piensa que algunos o todos los genes que confieren la actividad hemolítica a la bacteria contribuyen en las interacciones patógeno-tejido del hospedero y que las especificidades y tipos de hemolisinas con respecto a la membrana de su célula blanco son importantes en las infecciones por H. pylori en humanos y a su estadío. (24). β-catenina Es una molécula que puede estar unida a la membrana y es un componente de las uniones adherentes basadas en cadherinas o libre en el citoplasma, siendo componente de la vía celular Wnt; la unión de Wnt a su receptor (Frz) inhibe la fosforilación de la β-catenina, lo cual provoca una acumulación nuclear de la misma que conlleva a la transcripción del Factor Amplificador de células T y a la activación de genes implicados en la carcinogénesis (25). La acumulación nuclear de β-catenina está aumentada en lesiones gástricas precancerosas (26). La catenina también puede ser activada por contacto con antígenos bacterianos como CagA, pero los mecanismos que utiliza H. pylori para la acumulación nuclear y activación de la β-catenina no están bien estudiados. Cuando hay acumulación nuclear de catenina ocurre transcripción anormal de genes tumorales y mutaciones de otros que son antitumorales como p53, así mismo hay un aumento en la producción de linfocitos T que colaboran en el ataque hacia la bacteria produciendo inflamación local que puede conllevar a procesos carcinogénicos (27). Flagelos La motilidad hacia las células epiteliales gástricas es de vital importancia para la supervivencia de H. pylori. Esta función está asegurada por diferentes factores, incluyendo los movimientos en espiral por la presencia de 2 a 6 flagelos polares, cuyos filamentos son de dos tipos, codificados por FlaA y FlaB, las cuales están diseñadas para navegar por el espeso moco gástrico, a través de modificaciones eficientes de la matriz extracelular y capa mucosa, y disminución de la viscosidad lo cual permite la penetración bacteriana (28). Además la ureasa de H. pylori requiere níquel; un transportador de alta afinidad (nixA) necesario para la óptima actividad de la ureasa (29). El gen flagelar de H. pylori flbA modula la actividad de la ureasa disminuyendo la actividad por diferentes mecanismos: Disminuye la actividad del promotor de la enzima, aumentando el número de subunidades de la ureasa y también disminuye la expresión del promotor de nixA (30). Las cepas de H. pylori carentes de flbA no podrían colonizar la mucosa gástrica debido a su incapacidad para moverse (31). Los flagelos no son determinantes de patogenicidad directos de la bacteria, pero mediante mecanismos indirectos contribuyen con la patogénesis de la infección causada por H. pylori. Gastrina Se ha descrito que los pacientes con infección por H. pylori tienen incrementada las concentraciones séricas de gastrina en ayunas y post-pandrialmente, y que las concentraciones disminuyen entre 2 a 14 dias de iniciada la erradicación de la bacteria (32,33). La gastrina es otro factor implicado en la carcinogénesis relacionada con H. pylori. Esta hormona al ser segregada a la luz gástrica en respuesta a la bacteria, puede estimular el crecimiento de H. pylori y a las células G para que secreten más gastrina, bloqueando la expresión del gen p21 y sobreexpresando la proteína p53 mutada (5). Varios estudios han demostrado concentraciones reducidas de somatostatina dentro de la mucosa antral en sujetos con H. pylori, hallazgos que sugieren que la hipergastrinemia asociada a H. pylori es secundaria a la pérdida del control inhibitorio ejercido por la somatostatina (32,34). El mecanismo por el cual H. pylori resulta en una depleción de la somatostatina antral aun debe ser dilucidado (35). El péptido liberador de gastrina (Bombesina) tiene acciones biológicas potentes, incluyendo efectos secretores y motores en el aparato digestivo. Su relación con la infección por H. pylori se pone en evidencia por estudios que señalan que la hipergastrinemia asociada con H. pylori, es estimulada y creada por la acción de la bombesina, pues una vez erradicada la bacteria, los altos niveles de esta hormona vuelvan a la normalidad (36). Algunos estudios señalan que los niveles de colecistocinina-8 en la gastritis crónica por H. pylori contribuyen a la aparición de la hipergastrinemia y a los trastornos de la motilidad (35). El incremento de gastrina durante la infección por H. pylori tiene poder oncogénico ya que produce mutaciones en genes que son supresores de tumores, lo cual conlleva a transformación celular y mitosis acelerada. Resistencia a Antimicrobianos Las enfermedades asociadas con H. pylori usualmente involucionan o curan completamente después del tratamiento con antimicrobianos, lo cual depende de una variedad de factores como la dosis, la duración de la terapia y el nivel de resistencia de H. pylori (37). Estas terapias poseen algunas limitaciones como por ejemplo la presencia de efectos secundarios, la necesidad de terapias combinadas y la eficacia limitada, en particular debido al desarrollo de resistencia a antimicrobianos (38,39). La resistencia a nitroimidazoles es la forma más común de resistencia antimicrobiana en H. pylori (39). En países industrializados alrededor del 35% de las cepas de H pylori son resistentes a nitroimidazoles, mientras que en países en vía de desarrollo las tasas de resistencia para estos medicamentos son mucho más altas, y en algunas regiones prácticamente todas las cepas de H. pylori son nitroimidazol-resistentes. (40) Esta diferencia en la prevalencia puede deberse al uso común de metronidazol y otros nitroimidazoles en países en vías de desarrollo para el tratamiento de enfermedades parasitarias, mientras que en países desarrollados estos medicamentos son principalmente utilizados para el tratamiento de infecciones dentales y ginecológicas (41). Hasta finales del siglo XX la resistencia a penicilinas y tetraciclinas era muy rara en H. pylori aunque en la actualidad ha aumentado la resistencia a amoxicilina y tetraciclina, sobre todo en regiones donde estos antibióticos pueden ser obtenidos sin prescripción médica (42). Diversos estudios han concluido que la erradicación del H. pylori es de gran utilidad para la prevención del desarrollo de una neoplasia gástrica (43). Así como han demostrado que la erradicación H. pylori esta asociada con una disminución en la tasa de recurrencia, en pacientes con resección endoscópica de estadios tempranos de cáncer (44) Algunos reportes hacen énfasis en la importancia del tratamiento contra esta bacteria en edades tempranas, de allí la importancia de evitar un ascenso en la resistencia bacteriana ya que se convertiría en un serio problema si continúa en aumento (40). Proteínas de shock térmico Son una familia de proteínas inducida en células eucariotas y procariotas bajo condiciones de estrés. Estas proteínas actúan como chaperonas al facilitar el plegamiento y translocación de polipéptidos intracelulares (45). La proteína del shock de calor de 60 Kd (HSP60), es un potente antígeno inmunológico de H. pylori (46) que induce la secreción de IL-8 y la expresión del ARNm de esta citocina por las células epiteliales gástricas humanas (47). HSP60 juega un rol en la adherencia de H. pylori al epitelio gástrico y es considerado un factor de virulencia bacteriano que puede inducir respuesta inflamatoria en el hospedero (48). Se ha comprobado que la respuesta humoral de HSP60 esta relacionada con inflamación gástrica y juegan un papel en la patogénesis del linfoma MALT (49). Algunos estudios han demostrado que receptores Toll-like (TLR) actúan como receptores para estructuras bacterianas como lipopolisacárido y HSP60. Dentro de la familia de los receptores TLR, TLR2 y TLR4 son los más importantes en el reconocimiento de HSP60 en células humanas. TLR2 y TLR5 son requeridos para que H. pylori induzca la activación del NF-kB y la expresión de quimiocinas por las células epiteliales (50). Recientemente, fue reportado que el HSP60 de H. pylori puede mediar la producción de IL-6 por los macrófagos por vías independientes de TLR (51). Al mediar la producción de ciertas citocinas como IL-6 e IL-8, la HSP60 colabora en el proceso de producción de anticuerpos por parte de los linfocitos B y con el proceso de inflamación gástrica, ésta inflamación al volverse crónica puede causar daños en los tejidos como úlceras que evolucionan en ciertas ocasiones a neoplasias. Catalasa Es una de las enzimas producidas por la bacteria que desempeña una función importante como factor de virulencia, favorece la sobrevivencia de la bacteria en el tejido inflamado, la protege de las acciones fagocíticas de los neutrófilos, de los metabolitos reactivos de oxígeno (MRO) y la de otros mediadores químicos de la inflamación. (17) Mucinasa Enzima proteasa que desintegra la estructura glicoproteíca del mucus y debilita su función como barrera por la pérdida gradual de su viscosidad, lo cual aumenta la retrodifusión del ion hidrógeno, y la capacidad de la bacteria para desplazarse en la mucosa gástrica, por ello es considerado un factor de mantenimiento ya que le permite a la bacteria colonizar y persistir en la mucosa del estómago (17,52). Lipasa y Fosfolipasas A2 Y C Estas sustancias, liberadas por la bacteria en el sitio de la lesión, son capaces de degradar los fosfolípidos del mucus y disminuir su hidrofobicidad, como consecuencia de su fuerte actividad lipolítica, de ahí su importancia en la ulcerogénesis. La lipasa y las fosfolipasas A2 y C, al generar lisofosfolípidos provistos de actividad lítica, pueden atacar la integridad de la membrana epitelial y favorecer la liberación de ácido araquidónico (AA), con la consiguiente producción de leucotrienos y otros eicosanoides que contribuyen a la inflamación. Estos compuestos, además de su acción inflamatoria, también alteran la permeabilidad de la membrana celular y la regeneración del mucus. (17) Superóxido Dismutasa Enzima que se encuentra en altas concentraciones dentro del citoplasma del H. pylori, utilizada por dicho microorganismo como mecanismo de defensa contra el ataque fagocítico de los neutrófilos. Actúa como antioxidante al catalizar los metabolitos reactivos de oxígeno producidos por los neutrófilos, que pudieran dañarla. (17) Factor Activador de Plaquetas La bacteria es capaz de sintetizar y liberar cantidades importantes del factor activador de plaquetas, con potente acción quimiotáctica sobre los neutrófilos y los eosinófilos, así como otras acciones inmunomoduladoras, incluyendo la proliferación de los linfocitos. Este factor es conocido también como un agente proulcerogénico en la mucosa gástrica por su acción sobre la adherencia y activación de los neutrófilos. (17) Lipopolisacáridos Numerosos trabajos reportan que la membrana externa que cubre la bacteria, es capaz de actuar como material antigénico y estimular la respuesta inflamatoria. (52) La membrana externa de la bacteria es rica en lipopolisacáridos, que son proteínas heterogéneas con baja actividad biológica, capaces de activar los monocitos y los neutrófilos; éstos, a su vez liberan citocinas, eicosanoides, metabolitos reactivos de oxígeno, activan el complemento en el sitio de la lesión y perpetúan la respuesta inflamatoria, como mecanismo de defensa ante los daños de la bacteria, que al producir más lipopolisacáridos provoca lesión hística local y síntomas sistémicos (fiebre), por lo cual los lipopolisacáridos constituyen uno de los principales antígenos del H. pylori. (17) Persistencia La acidez gástrica y el peristaltismo son mecanismos para evitar la colonización bacteriana del estómago, pero H. pylori tiene diversos mecanismos para evadir las defensas primarias del hospedero y establecer una infección persistente. Algunas cepas poseen BabA2 lo cual le confiere al susceptible un alto riesgo de padecer lesiones precursoras de cáncer gástrico. (53) La adherencia de H. pylori puede ser finamente modulada, lo cual es importante tanto para la colonización inicial como para la persistencia bacteriana. H. pylori posee tropismo celular estricto, colonizando la mucosa gástrica de humanos y primates. Incluso hay ciertas regiones dentro del estómago donde se puede evidenciar aún más la presencia de un mayor número de bacterias como ocurre en las glándulas gástricas de la parte alta del estómago (54). Para la exitosa persistencia de la bacteria en la mucosa gástrica, esta de ser capaz de evadir la respuesta inmune del hospedero. Las especies reactivas de O2 y nitritos que pueden producir daño oxidativo al ADN son generadas por la presencia de neutrófilos presente en la mucosa inflamada. Estas especies reactivas no solo dañan al tejido del hospedero, pero adicionalmente pueden dañar a los microorganismos allí presentes, es por eso que se dice que H. pylori puede sobrevivir bajo condiciones de estrés oxidativo (55). En modelos animales y humanos, la infección con H. pylori lleva a una gran producción de IgG e IgA, ambos presentes en la mucosa gástrica y en el suero. Usando un modelo de ratón deficiente en células B fue demostrado que la colonización inicial era comparable con aquella observada en el ratón salvaje, la bacteria fue completamente eliminada del ratón mutante dentro del contexto de inflamación gástrica severa. En contraste, el ratón salvaje permaneció colonizado durante el estudio pero desarrollo solo gastritis leve. Esto sugiere que la presencia de anticuerpos da como resultado una inflamación menos severa, pero conlleva a una colonización persistente (56). A pesar de las estrategias utilizadas por H. pylori para evadir la respuesta inmune, una activación inmune significante aun ocurre. Estudios recientes señalan que el H. pylori puede utilizar sus factores de virulencia para regular y evitar los efectos inmunitarios. La isla de patogenicidad Cag codifica un sistema de secreción tipo IV el cual introduce productos bacterianos a las células del hospedero; tales como CagA, peptidoglicanos, entre otros (57). Un factor adicional relacionado con la patología de H. pylori es VacA, una citotoxina bacteriana. VacA puede contribuir a la evasión de la respuesta inmune adaptativa (58). En contraste con CagA, VacA bloquea la activación de NFAT, y estudios recientes indican que VacA inhibe la expansión clonal de los linfocitos T CD4+ dependiente de NFAT (59). Esto contribuye a la capacidad de H. pylori de evadir la respuesta inmune y establecer una colonización persistente (51). Factores que influyen en el desarrollo de la respuesta inmunitaria y participan en laoncogénesis Antecedentes genéticos El cáncer gástrico se asocia con el H. pylori y sus factores de patogenicidad; sin embargo, los avances en el estudio del cáncer han demostrado que el hospedero tiene antecedentes predisponentes como la carga y el polimorfismo genético que se va a ver reflejado en la regulación de la respuesta inmunitaria en contra de la bacteria. Recientemente, el polimorfismo genético del gen de la interleucina -1β (IL-1β) y del gen del antagonista del receptor de la misma (IL-1RN), han sido asociados con el incremento del riesgo a padecer hipoclorhidria y carcinoma gástrico. La IL-1β es una citocina con un potente efecto proinflamatorio que también tiene la capacidad de inhibir poderosamente la secreción de ácido gástrico, y juega un gran papel en el inicio y amplificación de la inflamación, en respuesta a la infección por H. pylori (60). Un alelo polimórfico con una timina (T) en lugar de una citosina (C) en la posición -511 de la región de regulación del gen de la IL-1β (IL-1β -511T) está asociado con un incremento en la producción de IL-1β. Por otro lado, el gen IL-1RN codifica la citocina antagonista del receptor de IL-1β, que se une de manera competitiva a dicho receptor, modulando así los efectos potencialmente dañinos de la IL-1β, por lo que tiene efecto antiinflamatorio. Este gen tiene un número variable de repeticiones en el intrón 2, teniendo así un alelo corto de sólo 2 repeticiones (IL-1RN2) y uno largo de 3 a 6 repeticiones (IL-1RNL). El gen con el alelo polimorfo IL-1RN2 esta asociado con el incremento de la producción de IL-1β (60). La infección por H. pylori en individuos con estos alelos (IL-1β -511T y IL-1RN2) puede tener como resultado un incremento en la producción de IL-1β gástrica, teniendo como consecuencia una inflamación crónica severa, atrofia gástrica, hipoclorhidria y por último puede desencadenar el desarrollo de un carcinoma gástrico (61). Existen muchos otros factores genéticos que favorecen la oncogénesis a nivel gástrico, que son predisponentes exista o no la infección por H pylori, pero que aunadas a la misma aumentan considerablemente el riesgo a padecer cáncer gástrico; como lo son las mutaciones en el gen de la E-Caderina en familias con cáncer gástrico hereditario de tipo difuso (62). Se estima que 8-10% de los carcinomas gástricos están relacionados a un componente familiar. El cáncer gástrico se ha observado en pacientes con poliposis gastrointestinal como pólipos adenomatoso familiar y Peutz-Jeghers; el incremento de riesgo de cáncer asociado con poliposis adenomatosa ha sido reportado en regiones de alto riesgo como Asia (62). Respuesta inmune (Reclutamiento de linfocitos) Las células Th1 generan una respuesta inflamatoria y de inmunidad mediada por células por la producción de INF-γ, IL-2, IL-12, IL-18 y TNF-α, una respuesta usualmente eficaz contra microorganismos patógenos intracelulares, mientras que las células Th2 son asociadas preferentemente con la inducción de anticuerpos IgA mediante la producción de IL-4, IL-5, IL-6 IL-10, IL-13 , entre otras citocinas. Siendo H. pylori un microorganismo extracelular, debido a que su hábitat lo constituye la interfase mucosal gástrica, la infección por esta bacteria modifica la diferenciación de las células Th0 hacia una respuesta inmune dominada por una respuesta inflamatoria tipo Th1. Aunque aún no es claro cómo ocurre esta modulación de la respuesta inmune, el proceso inflamatorio y la respuesta inmune celular contribuyen significativamente a la patología gástrica asociada a la infección por H. pylori (27). Sin embargo, esta respuesta con Th1 con altísimos títulos de IgM e IgG (dependiendo del tiempo de la infección), no es protectora y clínicamente es apenas un marcador de enfermedad y no de inmunidad. Por la importancia que tiene actualmente la infección por H. pylori y la afirmación indiscutible de que es el agente etiológico más común de la inflamación gástrica, describiremos los diferentes procesos que ocurren tras su llegada al estómago. La patogénesis de la gastritis crónica por H. pylori incluye dos etapas:
Enfermedades de base Varios metaanálisis han sugerido que la infección crónica por H. pylori induce a un incremento dos o tres veces superior en el riesgo de desarrollar cáncer gástrico. La patogénesis no está clara, pero el incremento en la proliferación celular de las células de la mucosa gástrica asociado con un reflujo biliar crónico parece ser un factor carcinogénico importante. La colonización e infección subsecuente por H. pylori en pacientes sometidos a gastrectomía parcial por enfermedad ulcerosa benigna no parece verse influida por el reflujo biliar, y por tanto, la bacteria podría tener un papel en la carcinogénesis gástrica de los pacientes gastrectomizados (63). Por otro lado, se encuentra la enfermedad de Menetrier, que es una gastropatía de aparición excepcional expresada macroscópicamente como un engrosamiento marcado de pliegues, y microscópicamente con hiperplasia de la superficie mucosa de las células foveolares, con criptas grandes y tortuosas, cuya causa permanece desconocida. En este sentido, se ha descrito recientemente una elevada prevalencia, en un 90 %, de infección por H. pylori en los pacientes que padecen dicha enfermedad, por lo que se ha sugerido que pudiera representar, al menos en un subgrupo de enfermos, una forma especial de gastritis causada por H. pylori (62). Reinfección La erradicación de H. pylori cura la úlcera activa y, a largo plazo, da lugar a una drástica reducción de la reaparición de la misma y sus complicaciones (64). Una vez erradicada la bacteria tras un tratamiento efectivo, existen dos tipos de recurrencia de la infección. Un mecanismo sería la recrudescencia, definida como la detección de una cepa de la bacteria similar a la aislada previamente a la erradicación. El otro mecanismo es la verdadera reinfección, esto es, cuando el paciente es infectado por una cepa distinta de H. pylori (65). Para diferenciar recrudescencia de reinfección se requiere la detección de distintas cepas bacterianas, bien mediante la reacción en cadena de la polimerasa (PCR) o el análisis de polimorfismos genéticos (RFLP) (66). Se han propuesto distintos factores como coadyuvantes de la recurrencia de la infección, como la falta de inmunidad natural para la reinfección o el bajo nivel socioeconómico (67). Otros trabajos citan como factores de riesgo la presencia de H. pylori en la placa dental (68) o el reservorio de la infección en el ámbito familiar (69). Otro aspecto motivo de controversia son las implicaciones clínicas de la reinfección por H. pylori. Tras la reinfección pueden ocurrir diversas circunstancias: la reaparición de la infección con o sin síntomas pero sin lesiones, la recidiva ulcerosa con reaparición de la clínica y complicaciones (fundamentalmente con sangrado), y la recidiva de linfoma MALT gástrico, tras haber regresado histológicamente después de la erradicación de la bacteria (70). La recurrencia ulcerosa en pacientes reinfectados oscila en distintas series entre el 3 y el 60%, con un promedio de 20% (71). La mayoría de los trabajos demuestran, además, que la erradicación es más efectiva que el tratamiento con antisecretores de mantenimiento para prevenir recidivas y complicaciones ulcerosas (72). Todos estos aspectos plantean dudas sobre el manejo de los sujetos reinfectados, preguntándose algunos autores si debería seguirse una comprobación periódica y mantenida del estado de infección por H. pylori en estos pacientes y una nueva erradicación cuando estuviera indicado clínicamente, sobre todo, en casos de linfoma MALT o hemorragia ulcerosa (73). La prueba de aliento debería ser la prueba diagnóstica de primera elección para la detección de reinfección, por su nula invasividad y capacidad para explorar toda la cavidad gástrica; pero también puede compararse con otros métodos como la histología o el test de ureasa rápida (74). Se podría proponer en aquellos pacientes erradicados y asintomáticos, especialmente menores de 50 años el seguimiento mediante prueba de aliento anual, durante los tres primeros años, para detectar una posible reinfección, que se beneficiara de un retratamiento, antes de la aparición de complicaciones. En cambio, en los pacientes erradicados sintomáticos se debería realizar endoscopia digestiva alta diagnóstica. La menor edad de los pacientes y un valor más elevado en el test de aliento posterradicación son factores predictivos de reinfección. Por ello, es recomendable un seguimiento con pruebas de aliento anuales, sobre todo, en menores de 50 años, para la detección de reinfección y así evitar complicaciones ulcerosas ulteriores (75). La tasa de reinfección se sitúa en países subdesarrollados entre el 6,25% en algunas zonas de la población asiática y el 2,4% en ciertas áreas de la población india (76). Por el contrario, la tasa en países desarrollados es inferior al 1% (77). Factor de crecimiento epitelial La infección de la mucosa gástrica por H.pylori afecta directamente en el balance normal entre la proliferación celular y la apoptosis, desregulando su ciclo normal, que se presenta con una disminución de la apoptosis en sus primeros estadios (gastritis y atrofias gástricas) y un gran aumento en la proliferación celular en los estadios mas avanzados. Se ha descrito la relación entre la proliferación celular y los cambios genéticos originados en el epitelio gástrico, lo cual está asociado a diversos factores, tales como la liberación de gastrina, factores de crecimiento celular, mutación del gen p53,gen asociado a la apoptosis, que media la activación de una serie de factores relacionados con el aumento de la proliferación celular, como la interleucina 6, factor de crecimiento epitelial (EGF, por sus siglas en idioma inglés), factor de crecimiento similar al de la insulina (IGF, por sus siglas en idioma inglés) y el gen de resistencia a múltiples fármacos(MDR 1, por sus siglas en idioma inglés). Éste desequilibrio en la regulación de los genes que median la apoptosis, pueden evolucionar en última instancia al desarrollo de un tumor. El proceso degenerativo de la mucosa gástrica comienza con la inflamación, conduciendo a la destrucción de las glándulas gástricas, cuando esto ocurre y es reemplazado por otro tejido, se produce una pérdida de glándulas (atrofia), trayendo consigo una reducción en la producción de ácido, característica asociada con el desarrollo de cáncer gástrico. La infección por el H.pylori induce la liberación de radicales del oxigeno, siendo éstos derivados de la activación de neutrófilos, los cuales son factores dañinos de la mucosa gástrica. La protección de las células contra estos radicales del oxigeno (ROS, por sus siglas en idioma inglés), radica en las enzimas secuestradoras de estas especies, como lo son superóxido dismutasa, catalasa y glutatión peroxidasa. La inflamación causada por la infección por H. pylori, produce cambios en la expresión enzimática, como es el caso de las enzimas ciclooxigenasas (COX), que catalizan la conversión de ácido araquidónico a prostanoides como la prostaglandinas, los cuales protegen la mucosa gástrica de la apoptosis, mediante el aumento de la proliferación celular (30). Los factores de crecimiento (FC) son proteínas solubles presentes en el suero imprescindibles para la proliferación celular, por lo que la formación del complejo FC-rFC (factor de crecimiento y su receptor) es un evento importante en la actividad proliferativa. Las mutaciones del complejo FCrFc producen un estimulo proliferativo constante e independiente de los mecanismos regulatorios, lo que conlleva una hiperproliferación, caraterísticos de los cánceres gástricos (78). Dieta alta en sal y nitratos Se ha señalado que la ingesta a largo plazo de elevadas concentraciones de nitratos en alimentos secos, ahumados y salados puede estar relacionada con un mayor riesgo de cáncer gástrico (2). La sal no es un agente carcinógeno, sin embargo, en experimentos de animales tratados con soluciones salinas hipertónicas, se observó un incremento en la proliferación celular en la mucosa gástrica, resultando finalmente en una gastritis atrófica, por lo que puede actuar como agente promotor de la oncogénesis ya que los cambios proliferativos inducidos pueden aumentar los efectos de carcinógenos derivados de alimentos (79). Por ello, se ha postulado que el continuo consumo de altos niveles de sal puede traer como consecuencia una gastritis atrófica que subsecuentemente aumenta el riesgo de cáncer gástrico (79). Los nitratos por su lado actuarían como agentes carcinogenéticos y mutagénicos, a través de la formación, en la mucosa gástrica de nitrosaminas y nitrosamidas (nitritos). La transformación de nitritos a nitratos se produciría por acción reductora de las bacterias. Las nitrosaminas determinarían modificaciones de la mucosa (metaplasia de tipo intestinal, gastritis atrófica), precursoras de la alteración neoplásica (80). Disminución de vitamina C Evidencia epidemiológica sugiere una fuerte asociación entre la alta ingesta de ácido ascórbico y la baja incidencia del cáncer gástrico. En estómagos normales la concentración total de vitamina C en el jugo gástrico es cinco veces la del plasma, aparentemente debido a una secreción activa de la mucosa gástrica, existiendo en dos formas principales: ácido ascórbico y ácido dehidroascórbico (forma más activa), el mayor porcentaje esta en la forma reducida. La protección que brinda el ácido ascórbico a la mucosa gástrica es de dos maneras: primero, depurando los radicales libres de oxigeno protegiéndola contra insultos oxidativos, y segundo, inhibiendo la formación de compuestos N-nitrosos por depuración de nitritos libres (81). H. pylori disminuye la cantidad de vitamina C en el jugo gástrico, a través de un mecanismo poco definido, pero se correlaciona con el grado de severidad de infiltración de polimorfonucleares en la mucosa antral, reduce además la proporción de su forma reducida activa; por otro lado se ha visto que en un pH neutro se encuentran reducidos los niveles de ácido ascórbico en el estómago, probablemente debido a que la bacteria produce una depleción del ácido ascórbico por producción de nitritos (82). La gastritis causada por la infección crónica por H. pylori resulta en hipoclorhidria, una condición que permite el crecimiento de diversas bacterias; las bacterias que son reductoras de nitrato pueden convertirlo en nitritos y luego en compuestos nitrosados. La inflamación causada por H. pylori ocasiona producción excesiva de especies reactivas del oxígeno que causan daños en el ADN mediante translocaciones y delecciones (17). Hipoclorhidria La hipoclorhidria debida al desarrollo de gastritis atrófica, o en parte funcional, puede favorecer el crecimiento de bacterias en el lumen gástrico; esta sobre población da lugar a altos niveles de nitritos en el lumen del estómago, que al combinarse con aminas y amidas originan compuestos N-nitroso. La saliva deglutida parece ser una fuente de nitratos importante en la generación de estos compuestos. Estos compuestos N-nitroso, o una acción directa del H. pylori (en una infección de largo término) podrían ser responsable de lesiones premalignas, metaplasia intestinal y displasia en la mucosa gástrica. Se ha observado que tanto la infección aguda como la crónica por H. pylori, pueden resultar en hiposecreción ácida marcada (81). Alcohol Se ha descrito una relación entre el consumo del alcohol y la infección por el H. pylori, aunque los estudios no han sido concluyentes, se encontró que el consumo de alcohol podría tener un efecto, mas que protector en contra de la infección, de la eliminación de la bacteria. Sin embargo, el estudio también sugiere que el consumo prolongado de bajas cantidades es factor determinante para que este efecto benéfico de eliminación pueda darse, siendo esta ingesta referida preferiblemente a vino tinto (83). Por el contrario, estudios realizados en Moscú arrojan datos totalmente distintos que develan que el consumo de bebidas alcohólicas fuertes, como el vodka, constituye un factor de riesgo elevado en el desarrollo de cáncer gástrico, aunque se observó una mayor tendencia en los hombres a presentar cáncer de cardias en comparación con las mujeres (84). Las diferencias entre los resultados obtenidos en los estudios realizados, en este tópico en particular, podrían basarse en el sesgo de la selección de las personas estudiadas, ya que los investigadores no tomaron los mismos parámetros en el momento de la selección del individuo a estudiar, lo cual es una hipótesis que manejan los autores. Tabaco Se ha demostrado mediante metaanálisis que el fumar aumenta el porcentaje de dificultades para el tratamiento de erradicación contra H. pylori (61). Varios factores tales como la resistencia a los antibióticos; la enfermedad del paciente, por ejemplo, úlcera péptica y el consumo de café han influido en la respuesta a la terapia de supresión de H. pylori. Un gran número de estudios han proporcionado evidencia de que el fumar obstaculiza la eliminación de H. pylori; sin embargo, varios artículos no encontraron ninguna asociación (61). Es importante entender los posibles mecanismos detrás de los efectos negativos del tabaquismo en la erradicación de la bacteria. En primer lugar, que fumando el flujo gástrico y la secreción de moco disminuyen y así, puede ser que se reduzca la entrega de antibióticos a la mucosa gástrica. En segundo lugar, el fumar estimula la secreción ácida, que se ha asociado al tratamiento debido a que la amoxicilina es un antibiótico ácido-sensible, y su eficacia puede disminuir en fumadores. En tercer lugar, el fumar puede modular la actividad de las isoenzimas específicas del citocromo p450 implicados en el metabolismo de inhibidores de la bomba de protones. En este sentido, el fumar puede conducir a la progresión de la gastritis atrófica y metaplasia intestinal en los pacientes infectados por H. pylori. Se encontró también que la nicotina tiene la capacidad de reforzar y potenciar la actividad de la toxina VacA de H. pylori en las células gástricas. Los fumadores infectados con H. pylori tienen 11 veces mayor riesgo de cáncer gástrico que los no fumadores no infectados (61). CONCLUSIONES Se puede establecer la relación existente entre la neoplasia gástrica y la infección por H. pylori, la cual es la bacteria responsable de la mayoría de los casos de gastroenteritis, úlcera péptica y gastritis crónica. En los individuos infectados, generalmente están presentes anticuerpos frente al H. pylori, pero no evitan la colonización posterior. Por ello, las personas que adquieren esta bacteria tienden a padecer enfermedades crónicas y asintomáticas por un largo período de tiempo. Una característica de la infección por este microorganismo en humanos es la gastritis, la cual puede persistir por décadas y evolucionar a cáncer gástrico o linfoma MALT, todo esto aunado a factores dependientes del medio ambiente, del hospedero y de la bacteria per se, tales como sus múltiples determinantes de patogenicidad. Por último, H. pylori es capaz de interferir en procesos fisiológicos protectores del estómago y duodeno, como la secreción de ácido gástrico, ácido ascórbico, gastrina y somatostatina; así como promover la apoptosis celular y la acumulación de factores que pueden provocar cambios en el genoma de la célula gástrica; todos estos fenómenos predisponen al cáncer gástrico, siendo entonces la erradicación de la bacteria una medida preventiva adecuada. Referencias
NOTA: Toda la información que se brinda en este artículo es de carácter investigativo y con fines académicos y de actualización para estudiantes y profesionales de la salud. En ningún caso es de carácter general ni sustituye el asesoramiento de un médico. Ante cualquier duda que pueda tener sobre su estado de salud, consulte con su médico o especialista. Copyright 2007 - Centro de Análisis de Imágenes Biomédicas Computarizadas CAIBCO, Instituto de Medicina Tropical – Facultad de Medicina, Universidad Central de Venezuela The following images related to this document are available:Photo images[va07026f2.jpg] [va07026f1.jpg] |
| |||||||||
{kind=link}
{kind=link}