 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
VITAE Academia Biomédica Digital, Número, No. 36, Julio-Diciembre, 2008 Genética Enfermedad de Fabry: Desde el Aminoácido a la Clínica Fabry's disease: From aminoacid to clinical manifestations J Villalobos1, V L Colina2 1Cátedra de Fisiología, Escuela de
Medicina Luis Razetti, Universidad
Central de Venezuela. Sección de
Investigaciones Cardio-Renales, Instituto
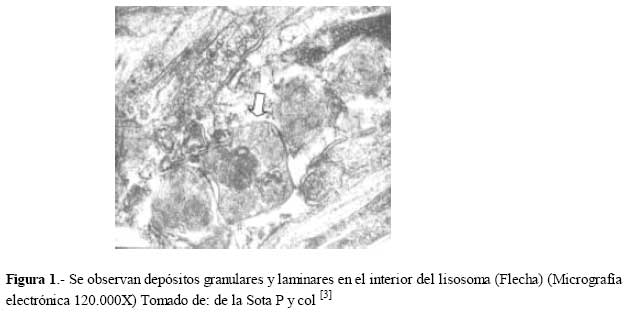
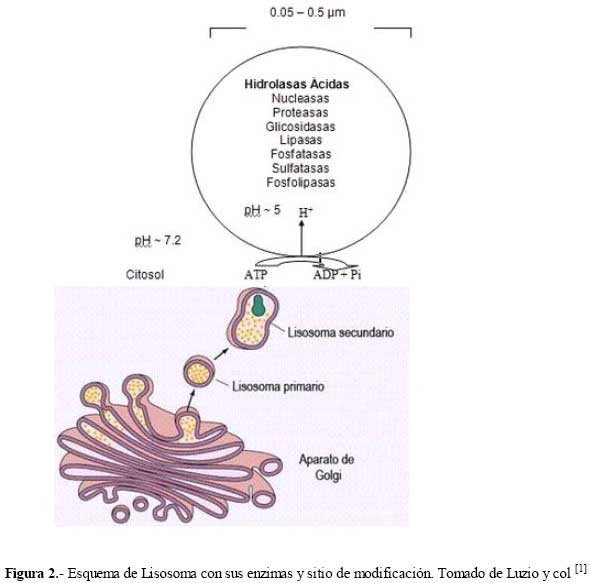
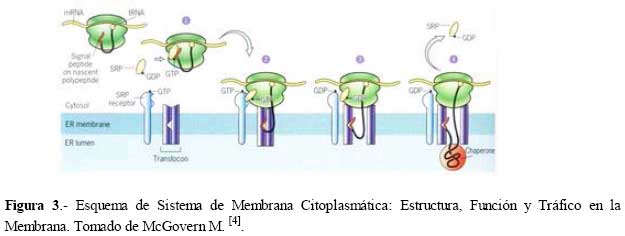
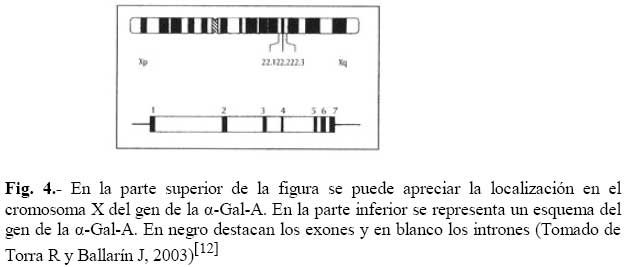
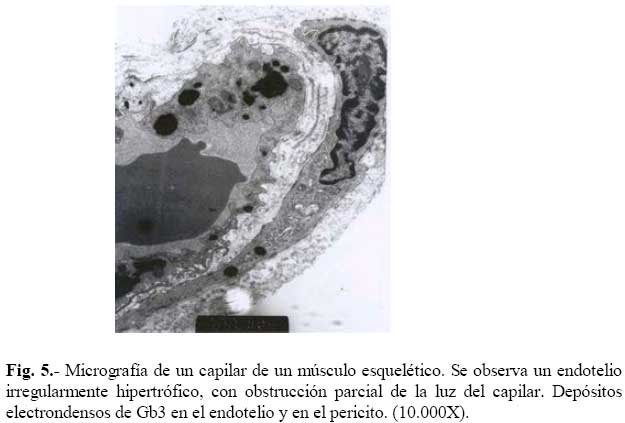
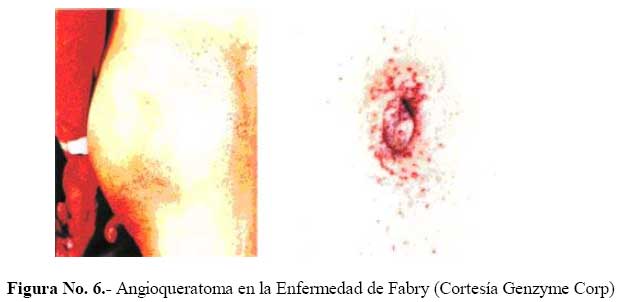
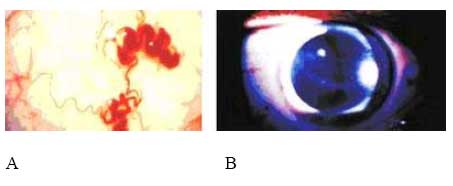
de Medicina Experimental Fecha de recepción: 11/11/2008 Code Number: va08020 Las Enfermedades de Depósito Lisosomal (EDL) constituyen un grupo de patologías en las que es indispensable el abordaje de las Ciencias Básicas para su comprensión, y las mismas son un ejemplo que rompe con la dicotomía aún no entendida entre las Ciencias Básicas y las Ciencias Clínicas, puesto que permite contemporizar entre ambos mundos, mutuamente incluyentes; es por ello, que con este trabajo se trata de dar una visión amplia de la Enfermedad de Fabry, haciendo un recorrido por sus características histomorfológicas, fisiológicas, fisiopatológicas y terapéuticas, que permitan alcanzar el análisis integral de esta entidad nosográfica. La Enfermedad de Fabry es una endoteliopatía de origen enzimático, con compromiso multisistémico. Es una Enfermedad de abordaje multidisciplinario, para la pesquisa, el diagnostico, tratamiento y pronóstico de la Enfermedad. Actualmente la terapia de reemplazo enzimático es una realidad con resultados satisfactorios que mejoran la calidad de vida de los pacientes y sus familiares. Palabras Claves: lisosomas, enfermedad de Fabry, terapia de reemplazo enzimático Abstract Lysosomal storage diseases (LSD) are a group of diseases in which knowledge of basic science is fundamental for understanding their pathology, and are an example that breaks the dichotomy between basic and clinical science. This work seeks to give a broad view of Fabry's disease, including: histomorphological, physiological, pathophysiological and therapeutic features, hoping to achieve a comprehensive analysis of this nosographic entity. Fabry's Disease is an endoteliopathy with enzymatic origin and multisystemic compromise. This disease requires a multidisciplinary approach for screening, diagnosing, treatment and prognosis of the disease. Currently enzyme replacement therapy is a reality with satisfactory results that improve the quality of life of patients and their families. Key Word: lysosome, Fabry disease, enzymatic replacement therapy Enfermedad de Fabry: Desde el Aminoácido a la Clínica Introducción La medicina moderna es el resultado de la sumatoria de esfuerzos en todas las áreas del conocimiento, en la que todas ellas participan aportando la información necesaria, que nos permita entender al individuo enfermo en toda su magnitud, desde lo absolutamente tangible hasta lo intangible; pero creíble según nuestra orientación personal, por lo que cada día deberíamos estar más capacitados para conocer al SER HUMANO en su máxima expresión. La clínica no es más que la resultante de la expresión de todos y cada uno de los procesos celulares y subcelulares, que actuando de manera sincrónica dan un todo que puede ser objetivo y cuantificable, por lo que en medida que se conozca más y mejor a cada uno de los procesos y mecanismos fisiológicos o fisiopatológicos podremos tener más y mejor información de lo que ocurre en el paciente; es por ello que el estudio de las Ciencias Básicas cada día debería ocupar mayor espacio en el concierto del conocimiento médico, para que la Medicina sea cada día más profesional y menos empírica. Las Enfermedades de Depósito Lisosomal (EDL) constituyen un grupo de patologías en las que es indispensable el abordaje de las Ciencias Básicas para su comprensión, y las mismas son un ejemplo que rompe con la dicotomía aún no entendida entre las Ciencias Básicas y las Ciencias Clínicas, puesto que permite contemporizar entre ambos mundos, mutuamente incluyentes; es por ello, que con este trabajo se trata de dar una visión amplia de la Enfermedad de Fabry, haciendo un recorrido por sus características histomorfológicas, fisiológicas, fisiopatológicas y terapéuticas, que permitan alcanzar el análisis integral de esta entidad nosográfica . I.-¿Que son los Lisosomas? En el citoplasma de las células animales se encuentran dispersos pequeños sacos de enzimas digestivas denominados lisosomas, los cuales son muy importantes para el catabolismo y el reciclado de macromoléculas dentro de una célula. Los lisosomas fueron descubiertos hace más de 50 años, como resultado de la asociación entre un número de hidrolasas, siendo una organela con propiedades definidas de centrifugación. Estas hidrolasas mostraron una latencia relacionada a la estructura, la cual depende de la integridad de la membrana que las rodea. Desde un inicio se reconoció que los lisosomas mostraban una morfología heterogénea, conteniendo depósitos densos y una membrana espiralada. (Fig. No. 1). En las células de mamíferos, se observan como organelas de ~ 0.5 µm de diámetro, frecuentemente con un núcleo electrón-denso, los mismos pueden llegar a ocupar entre 0.5 a 5 % del volumen celular y se concentran cerca de los microtúbulos, que se organizan en el centro de la célula. Los lisosomas han sido considerados como el compartimiento terminal de degradación de la vía endocítica y que de acuerdo al tamaño de las partículas ingeridas, dicha vía puede ser dividida en: pinocitosis (vesículas < 150 nm) y en fagocitosis (partículas > 250 nm); incluso participan en la autofagia, crinofagia y proteolisis de algunas proteínas citosólicas transportadas a través de la membrana lisosomal. La consideración del lisosoma como una simple “unidad de depósito de basura” ha cambiado recientemente, debido a un mejor entendimiento de cómo se entrega el material endocitado al lisosoma e incluso por la identificación de lisosomas que secretan su contenido después de unirse a la membrana plasmática [1]. Un lisosoma típico contiene más o menos 50 enzimas diferentes que degradan moléculas complejas, incluyendo lípidos, proteínas, carbohidratos y ácidos nucléicos, que se originan dentro y fuera de la célula, entre dichas enzimas se encuentran: proteasas, lipasas, fosfolipasas, glucosidasas, fosfatasas, sulfatasas, nucleasas; estas enzimas son producidas en el retículo endoplasmático rugoso (RER) y son posteriormente modificadas en el Aparato de Golgi (AG), en donde se identifican y distribuyen en los lisosomas (Fig. 2). Las moléculas de bajo peso molecular producto de la digestión enzimática pueden ser transportados a través de la membrana lisosomal al citosol. Las enzimas lisosomales exhiben una propiedad importante: todas tiene actividad óptima a pH ácido (pH=4.6), por lo que son hidrolasas ácidas [1,2]. La alta concentración interna de protones se mantienen por un constante transporte de protones (H+-ATPasa) presente en las membranas de la organela (Fig. 2). La membrana lisosomal contiene una variedad de proteínas integrales ácidas altamente glicosiladas, que probablemente la protegen del ataque por parte de las enzimas que la constituyen [2]. Las enzimas lisosomales son sintetizadas en el RER, usualmente como formas precursoras que sufren procesos post- translacionales (fragmentación proteolítico, la adición de oligosacáridos en el extremo N-terminal y la síntesis de marcadores de reconocimiento, como la manosa-6-fosfato), previo a formar los lisosomas primarios. Estos lisosomas se fusionan con otras vesículas, incluyendo fagosomas y autofagosomas que contienen macromoléculas, detritus celulares y organelas celulares, para formar lisosomas secundarios donde ocurre la degradación de las moléculas. Mediante la endocitosis, los lisosomas actúan en la captación de la vitamina B12, lipoproteínas, hormonas peptídicas y factores de crecimiento [4]. El proceso de síntesis de las enzimas lisosomales comienza cuando el RNAm se une a un ribosoma libre, comienzan a ser sintetizadas las enzimas lisosomales. Los polipéptidos ensamblados en los ribosomas unidos a la membrana, contienen una secuencia señal, de 6 a 12 residuos de aminoácidos no polares, que llevan al polipéptido naciente a la membrana del RE y permite la compartamentalización del polipéptido dentro de la luz del RE. Con frecuencia, el polipéptido señal se ubica cerca del extremo N-terminal. Una vez que sale del ribosoma, la secuencia señal es reconocida por una “partícula de reconocimiento de señal” (PRS), la cual está formada por 6 polipéptidos distintos y una pequeña molécula de RNA, llamada 7SL RNA. El PRS se enlaza tanto a la secuencia señal del polipéptido naciente como al ribosoma, impidiendo posteriormente la síntesis del polipéptido y previniendo plegamientos prematuros de residuos amino-terminales aberrantes. Se detiene la traslación, deteniendo el enlazamiento del PRS hasta que el complejo haga contacto y se unan a la membrana del RE. El PRS es una marcador que permite que todo el complejo (PRS-ribosoma-polipéptido naciente) se una a la superficie citoplasmática de la membrana del RE. La unión ocurre a través de por lo menos 2 tipos de interacciones: una entre el PRS y el receptor del mismo, y la otra entre el ribosoma y una proteína de la membrana del canal, llamada translocón. Una vez que el ribosoma se une firmemente a la membrana del RE, el asa terminal del polipéptido naciente es insertado dentro de un canal acuoso del translocón; dicha unión parece disparar un cambio conformacional que abre el canal a la luz del RE. En pasos posteriores, el PRS es liberado de su receptor en el RE, se reanuda la traslación, y el péptido es translocado a través del canal, a la luz del RE. Una vez terminada la translación, el ribosoma unido a la membrana es liberado, se completa el proceso y se cierra el canal de la membrana. Muchos de los pasos relacionados con la síntesis de proteínas secretoras y lisosomales, son regulados por la unión o hidrólisis de GTP. La interacción del PRS con un ribosoma parece inducir la unión de GTP al PRS, lo cual activa al PRS y le permite acoplarse con el receptor en la membrana del RE [2] (Fig.3). Las proteínas lisosomales sintetizadas en los ribosomas unidos a la membrana del RER, entonces son transportadas a la cisterna Cis del aparato de Golgi junto con otros tipos de proteínas. Una vez que se encuentran en las cisternas, las enzimas lisosomales solubles son reconocidas por enzimas que unen un grupo fosfato a la manosa de las cadenas de carbohidratos unidas al N-terminal. En el cis-Golgi se fosforilan uno o más residuos de manosa en el oligosacárido Man8(GlcNAc)2 mediante dos reacciones secuenciales. En la primera reacción, se añade un residuo de N-acetilglucosaminafosfato al átomo de carbono 6 de la manosa en el oligosacárido unido al amino terminal por acción de la N-acetilglucosamina fosfotransferasa, una enzima específica para las enzimas lisosomales. En la segunda reacción, el residuo de N-acetilglucosamina es removido por una fosfodiesterasa, dejando un residuo de manosa-6-fosfato (M6P)[5] . A diferencia de otras glicoproteínas distribuidas en la red trans-Golgi (TGN), las enzimas lisosomales poseen residuos de manosa fosforilados, que actúan como señales de reconocimiento. Las enzimas lisosomales que contienen esta señal son reconocidas y capturadas por receptores de manosa-6-fosfato (MPR), que son proteínas integrales de la membrana, concentradas dentro de una hendidura cubierta de clatrina del TGN . Se piensa que los receptores de manosa-6-fosfato atraviesan la membrana del TGN en diferentes sitios de unión, por lugares opuestos de la membrana. Mientras que una parte de los receptores de la manosa-6-fosfato, se proyectan hacia dentro de la luz del TGN, este se une a una enzima lisosomal, y una parte del mismo se proyecta hacia el citosol y se une a un complejo adaptador-clatrina que se enlaza a la superficie citosólica de la membrana del TGN. Estas interacciones aseguran que las enzimas lisosomales sean englobadas en vesículas cubiertas de clatrina, y que una vez liberadas al citoplasma, las mismas se dirijan a un destino particular que son los endosomas tardíos y posteriormente darán origen a los lisosomas. Los MPRs se disocian de las enzimas lisosomales antes de llegar al lisosoma, y los mismos son devueltos al TGN en vesículas sin cubierta. Los receptores de manosa-6-fosfato están localizados en la membrana plasmática, donde son capaces de atrapar enzimas lisosomales que son secretadas al espacio extracelular, y se reintegran a las enzimas en una vía que las devuelve a los lisosomas. La vía de distribución de la manosa-6-fosfato para las enzimas lisosomales señala varios principios importantes que se aplican a la distribución de las proteínas secretoras y de membrana. Primero, el residuo de manosa-6-fosfato es una de las señales de distribución que llevan a las proteínas a diferentes compartimientos dentro de la vía secretora. Segundo, los receptores de membrana difunden con sus ligandos dentro de discretas regiones de la membrana de una organela, donde son incorporados dentro de vesículas de transporte. Tercero, estas vesículas de transporte se unen solamente con una organela específica, el endosoma tardío. Y finalmente, los receptores de transporte celular, son reciclados después de que se disocian de sus ligandos [2]. ¿Qué son las Enfermedades de Depósito Lisosomal? Las Enfermedades de Depósito Lisosomal (EDL) son una familia de más de 30 desórdenes que resultan de diferentes defectos en la función lisosomal (Tabla 1). Los lisosomas degradan macromoléculas a sus constituyentes básicos: péptidos, amino ácidos, monosacáridos, ácidos nucleicos y ácidos grasos. Hers HG (1963) definió a las Enfermedades de Depósito Lisosomal como desórdenes caracterizados por deficiencia de una sola enzima lisosomal y la consecuente acumulación de su sustrato en los lisosomas [4]. Los glucoesfingolípidos (GSL) son componentes de las membranas plasmáticas que son degradados en los lisosomas después de su internalización a través de la vía endocítica. Su catabolismo requiere de la acción concentrada de varias enzimas hidrolíticas y de varios cofactores (como por ejemplo, las proteínas activadoras de esfingolípidos). La deficiencia de estas enzimas o de sus cofactores conlleva a distintas patologías clínicas tales como: la enfermedad de Tay-Sacks, Gangliosidosis, Fabry, Gaucher, todas ellas asociadas con manifestaciones características que reflejan los sitios tisulares específicos de depósitos de los sustratos parcialmente degradados. III.-Enfermedad de Fabry La enfermedad de Fabry es un error innato del catabolismo de glucoesfingolípidos, ligado al cromosoma X, debido a la deficiencia en la actividad de la exoglucohidrolasa α-galactosidasa A lisosomal (α-Gal-A). Afecta más a los hombres que a las mujeres: se calcula que 1 en 40.000 varones tienen la enfermedad de Fabry, mientras que la prevalencia en la población general es de 1 en 117.000 personas. Cuando una persona hereda el gene anormal que causa la enfermedad de Fabry, su cuerpo no puede producir suficientes cantidades de alfa-galactosidasa A o alfa-GAL[6]. Fisiopatología de la Enfermedad de Fabry Como resultado del defecto en la actividad de la hidrolasa lisosomal α-Gal-A, se acumulan sistemáticamente globotriaosilceramida y glucoesfingolípidos con residuo α-galactosil terminal, principalmente en los lisosomas del endotelio vascular y en el plasma [7]. Los GLS con residuos terminales α-galactosil, incluyen a los grupos sanguíneos B, B1, Pk y el antígeno P1; y los mismos se derivan principalmente del intercambio de las células, en los riñones, el hígado, los pulmones y los eritrocitos. El metabolismo endógeno es una gran fuente de sustrato en tejidos vasculares tales como la cornea, y las células neurales, las cuales están protegidas por la barrera hematoencefálica de los niveles circulantes elevados de ceramida trihexosa. Los GSL plasmáticos son sintetizados principalmente en el hígado e incorporados mediante la circulación sistémica, a partículas de lipoproteínas (LDL y HDL). Se ha calculado que el tiempo de intercambios de los GSL plasmáticos está entre 4 a 8 días, a una tasa de 1 a 6 µmol / día. Los estudios cinéticos sugieren que cada día se sintetizan de novo cerca de 25% de los GSL plasmáticos y una proporción de ellos deriva del intercambio de los eritrocitos senescentes [8]. La α-Gal-A nativa tisular humana, tiene un peso molecular de 100.000 dalton [9]. Su estructura fue determinada mediante métodos cristalográficos de rayos X, demostrando que está constituida por una glicoproteína homodimérica, con cada monómero compuesto por dos dominios, un dominio (β/α)8 que contiene el sitio activo, y un dominio C-terminal que contiene ocho cadenas β antiparalelas en dos hojas en un sándwich β. Después de la remoción de la secuencia señal de 31 residuos, el primer dominio se extiende desde los residuos 32 al 330 y contiene el sitio activo, formado por los extremos C-terminal de las cadenas β en el centro de la estructura en forma cilíndrica o de barril, una localización típica para el sitio activo en los dominios (β/α)8. El segundo dominio, comprendido entre los residuos 331-429, se pliega contra el primero en una extensa interfase. Visto en tercera dimensión, la molécula es cóncava ysu grosor varía entre 20 a 50 Å. El dímero está cargado negativamente y tiene entre un 7 y 15% de carbohidratos con un número variables de residuos de ácido siálico. Los grupos carboxilados están más concentrados alrededor del sitio activo, pero muchos de estos grupos se protonan, reduciendo la carga de la molécula, debido al pH tan bajo de los lisosomas [9,10]. La α-GAL humana se une a la α-galactosa haciendo uniones específicas con cada grupo funcional en el monosacárido. En humanos, la enzima hace contacto con cada grupo funcional en el ligando α-galactosa, y la misma muestra poca especificidad por la porción distal del sustrato más allá del ligando glucosídico, y se encuentra una hendidura activa en una amplia abertura sobre la superficie cóncava de la enzima [10]. Genética de la Enfermedad de Fabry Las células hepáticas humanas, sintetizan α-Gal-A como un precursor de peso molecular 58000 kD y es posteriormente procesado a una forma de peso molecular de 49000 kD. el ADN de la enzima humana ha sido clonado y codifica la secuencia proteica completa de la enzima madura [11]. En 1996, Miyamura y col.[7] ubicaron en el brazo largo del cromosoma X (Xq22.1) (Figura 4), la secuencia genómica de 14Kb y en el 2002, Pastores y col. [8] encontraron que el cromosoma contiene 7 exones que codifican el polipéptido α-Gal-A de 429 aminoácidos, incluyendo el péptido señal de 31 residuos en el extremo amino terminal. Las diferentes mutaciones en los genes estructurales lisosomales favorecen la heterogeneidad, que se observa en la expresión clínica de estas enfermedades. Por ejemplo, una mutación en un gen que codifica para una enzima lisosomal puede causar pérdida total de la actividad enzimática, mientras que otras mutaciones en el mismo gen, puede resultar en sólo una alteración parcial de la actividad enzimática, por consiguiente el curso clínico de la enfermedad sería menos severo. Otras mutaciones generan modificaciones post-translacionales o la afinidad a diferentes sustratos [4]. Aproximadamente el 57% de los alelos enfermos son mutaciones en donde un solo aminoácido es sustituido por otro (mutación missense), 11% sin sentido (mutación non sense), 18% supresiones genéticas parciales, 6% inserciones y 6% defectos en el procesamiento del RNA debido a uniones genéticas aberrantes. Se han encontrado mutaciones en los 7 exones. En la enfermedad de Fabry muchas de estas mutaciones son sustraídas y confinadas a una sola familia. El defecto en el gen α-GAL A, podría estar asociado en varios niveles, que estarían bien sea a un nivel de la proteína como tal o en la actividad enzimática indetectable (como resultado de una transcripción inestable de RNA), o con niveles perceptibles de la proteína enzimática, pero con actividad enzimática indetectable (mutaciones que afectan el sitio catalítico) y/o actividad de residuos de α-GAL A que se pueda medir (mutaciones que afectan el Holding proteico, la unión a sustrato o la tasa de recambio) [8]. La monosomía del cromosoma X se produce por una compensación de dosis, un sistema de todos los organismos con determinación sexual XY, la cual trae equilibrio de la expresión de muchos genes ligados al cromosoma X en mujeres y hombres. En mamíferos, este sistema de compensación incluye el “silenciamiento” de los genes en el cromosoma X el cual se conoce como Inactivación Cromosómica. En 1961 Lyon MF [12], planteó que en el desarrollo temprano, los cromosomas X en el embrión femenino, están inactivados al azar, por ello en algunas células de la madre, el cromosoma X se consideraba inactivado. Una vez que ocurría la inactivación en una célula, el patrón de inactivación del cromosoma X se transformaba en una parte fija de la herencia somática de las células. Por consiguiente, en las hembras se observaba como un mosaico, con algunas células expresando genes del cromosoma paterno y otras expresando genes del cromosoma materno [13]. En ratones, los dos cromosomas X del embrión femenino temprano no están diferenciados, tanto citológica como funcionalmente; ambos cromosomas X son activos. La inactivación en la línea germinativa es cíclica. En las hembras, un cromosoma X es inactivado durante la fase mitótica u oogonial, mientras que ambos cromosomas X son activados en la oogénesis. En los machos, debido a que tienen solo cromosoma X, no se presenta la inactivación en las células somáticas. La inactivación del cromosoma X en las células somáticas generalmente es al azar, teniendo la misma probabilidad de inactivación tanto con los cromosomas X materno como paterno [13]. El proceso de inactivación de los cromosomas se divide en tres fases:1.- Iniciación; 2.-Extensión, y 3.- Mantenimiento. 1.- Iniciación A partir de estudios de translocación y supresión del cromosoma X autonómico, surge la evidencia de la existencia y la localización de un centro de inactivación del cromosoma X (XIC), el cual interfiere con la manifestación de la inactivación del mismo. Un gen único que mapea para XIC, tiene propiedades particulares, sugiriendo que puede ser el locus esencial para el inicio de la inactivación del cromosoma X, y el mismo ha sido designado como Transcriptor Específico de la Inactivación de X (XIST). Esta es una fase heterocromática, de lenta replicación, con hipermetilación de las regiones promotoras resistente a nucleasa. La metilación de la citosina ocurre enzimaticamente después de la síntesis de DNA, y en mamíferos está restringida al dinucleótido 5 ´-CpG-3`. Esta mutación reprime la transcripción, y en el genoma en células normales, la gran mayoría de ellos se encuentran en el cromosoma X inactivo. Esto es importante en el mantenimiento del estado de represión de los genes ligados al cromosoma X inactivo, e incluso juega un papel importante en el establecimiento de la inactivación del cromosoma X [13]. Además, en esta fase ocurre una hipoacetilación (relacionada con el silencio genético) en los residuos de lisina en las histonas, enriquecida para la Histona Macro H2A; y muy importante, cubierta por XIST-RNA. Si el extremo 5’ del gen XIST es suprimido, el cromosoma no puede ser inactivado, mientras que la supresión de 3’ del gen, asegura la inactivación. Se ha demostrado que a pesar de que XIST se inicie en 5’, se transcribe un transcriptor antisentido, llamado TSIX, desde el promotor 3’ al locus XIST. Al igual que XIST, TSIX produce un RNA nuclear no traducido, y juega un papel importante en la protección del cromosoma de ser cubierto por XIST e inactivado (regulación de XIST). En las etapas embrionarias muy tempranas, XIST sólo se transcribe en el embrión femenino, y sólo del cromosoma X paterno. Por otro lado, TSIX se transcribe tanto en embriones masculinos o femeninos, y sólo del cromosoma X materno. En las células somáticas, la mutilación está incluida en el mantenimiento de un gen XIST silente en el cromosoma X activo; pero en el embrión temprano, la evidencia disponible indica que la mutilación no está comprometida con la expresión de XIST. Al mismo tiempo, en el que se está formando XIST-RNA en el embrión temprano, la histona macro H2A, ha mostrado asociarse preferencialmente con la inactivación del cromosoma X. 2.- Extensión. La extensión de la inactivación es un enigma. Se ha planteado que la extensión de la inactivación ocurre de una manera dominio por dominio, tal vez mediada por un locus de control llamado Región de Inactivación unida al cromosoma X. 3.- Mantenimiento. La mutilación y la replicación tardía son factores críticos para el mantenimiento del silenciamiento del cromosoma X inactivo [13]. En los estados de mórula tardía o blastocisto temprano, se observan evidencias tempranas del XCI: asincronía de la replicación del DNA entre los cromosomas, expresión génica diferencial y formación de la cromatina sexual (cromosoma X inactivo). En estas células, el cromosoma X paterno es inactivado preferencialmente. En el momento de la gastrulación ocurre la inactivación al azar del cromosoma X; una vez que ocurre la inactivación, toda la descendencia de una célula puede tener el mismo cromosoma X silente [13]. En ambos sexos, los genes responsables de las enfermedades ligadas al cromosoma X tienen riesgos clínicos diferentes. Debido a que las mujeres tienen 2 cromosomas X, deben ser heterocigoto u homocigoto para un gen mutante, y el gen alelo mutante puede demostrar expresión recesiva o dominante. En las mujeres, con frecuencia la expresión genética es variable e influenciada por la inactivación al azar del cromosoma X. Por otra parte, los hombres tienen solamente 1 cromosoma X, así que ellos son más propensos a mostrar un fenotipo completo. Por consiguiente, los términos “dominante ligado al cromosoma X, “recesivo ligado al cromosoma X” hacen referencia solamente a la expresión de las mutaciones en mujeres. En general, las enfermedades recesivas son muy raras, debido a que la capacidad biológica reducida de los homocigotos sirve para remover el gen mutante de la población [4]. Muchas de las deficiencias enzimáticas en las enfermedades recesivas, involucran enzimas de las vías catabólicas, normalmente las enzimas que degradan moléculas orgánicas de la dieta diaria, como son la galactosa, la fenilalanina y el ácido titánico (Síndrome de Refsum). Cuando la deficiencia de la enzima afecta una hidrolasa ácida (desórdenes de depósito lisosomal), el sustrato el cual usualmente es un lípido complejo o un polisacárido, el mismo se acumula dentro de los lisosomas edematizados. En las mujeres, la inactivación del cromosoma X al azar es el determinante más importante de la expresión de los desórdenes ligados al cromosoma X. Muchos individuos son asintomáticos, otros presenta síntomas moderados y otros tienen manifestaciones severas. La frecuencia de las alteraciones fenotípicas detectables, depende de un examen cuidadoso de los heterocigotos y de la edad en que se evalúen los mismos. La enfermedad de Fabry, junto con la Hemofilia A, la Distrofia Muscular de Duchenne, son ejemplos de enfermedades en las que pueda observarse expresión clínica en mujeres [4]. El análisis del gen de α-Gal-A, en la enfermedad de Fabry, mostró la existencia de lesiones moleculares heterogéneas, tales como mutaciones puntuales y reordenamiento genético parcial. Toshio y col (1999)[14] utilizando en ratones el modelo experimental de la enfermedad de Fabry, señalaron que los genes que codifican para α-Gal-A , son muy similares en tamaño, organización y secuencia nucléotidica de las regiones de codificación tanto en humanos como en ratones. Se han reportado más de 70 mutaciones en el gen que codifica para esta enzima, muchas de las cuales resultan en el fenotipo clásico de falta de actividad de la α-Gal-A [7]. Entre alguna de estas mutaciones se ha descrito una con diferentes codones de detención prematura, y todos los pacientes hemizigotos con dichas mutaciones, incluyendo E398X, manifiestan el fenotipo clásico de la enfermedad y probablemente solo fallan en 32 aminoácidos del extremo carboxilo terminal. Por otra parte se ha descrito que 26 de 28 residuos de aminoácidos deben ser eliminados proteolíticamente del extremo carboxilo terminal de la enzima galactosidasa, para generar el péptido final. Esto sugiere que la región carboxilo terminal debe ser uno de los extremos activos o debe tener influencia en la conformación del sitio activo [7]. Tanto clínica como bioquímicamente, la enfermedad de Fabry muestra una alta variabilidad fenotípica. Se podría observar heterogeneidad intrafamiliar, lo que se explica en pacientes con la misma mutación, por la presencia de genes modificados que cooperan en la expresión de α-Gal-A. Las mutaciones más frecuentes, descritas en la enfermedad de Fabry son las lesiones genéticas R227Q (con compromiso neurológico) y R220X, y se correlacionan con la forma clásica de la enfermedad. En el 2003, Morrone y col. [15] explicaron que su alta frecuencia en las mutaciones se debe a la presencia de CpG en el gen de α-Gal-A. El 93% de los genotipos está asociado con la enfermedad clásica, mediante mutaciones sin sentido y con una lectura de los codones en manera diferente, las cuales causan una terminación prematura de la síntesis proteica. Muchas mutaciones en donde un aminoácido es sustituido por otro, afectan los sitios catalíticos, sitios de dimerización o la forma intricada tridimensional de las proteínas [8]. Morrone y col, (2003) también observaron que las mutaciones que más se relacionan con daño renal son las S78X, C126-127, CATG y la A352D. Se observó en los leucocitos periféricos la presencia de α-Gal-A residual cuantificable, asociada con un manifestación tardía de la insuficiencia renal crónica, con un menor contenido de ceramida trihexosa y menor grado de daño histológico renal. Además se ha observado que las mutaciones conservativas donde un aminoácido es sustituido por otro estaban asociadas con una mayor sobrevida renal, comparada con las mutaciones no conservativas [8,15]. Sin embargo, se ha observado que en hombres algunas de esas mutaciones con sustituciones únicas de aminoácidos, localizadas en la región carboxilo terminal llevan a variantes atípicas de la enfermedad de Fabry, con manifestaciones limitadas al corazón. Se ha sugerido que esta variedad debe ser más común de lo que se piensa, dado que se presenta en pacientes masculinos con hipertrofia ventricular izquierda inexplicable [7]. Solo se han reportado 18 genotipos asociados con la variante cardiaca de la enfermedad de Fabry; y la mayoría de ellas son mutaciones en donde está involucrado un solo aminoácido. Muchas de esas mutaciones producen inestabilidad de la proteína, debido a un empaquetamiento inapropiado de la estructura proteica, y una alteración en el sitio de glicosilación (N215S). Han sido reportadas cuatro mutaciones (R112H, R301Q, G328R y R404) asociadas a la variante cardíaca en una familia y asociadas a Fabry clásico en otra [7]. No están completamente claras, las bases para las variaciones fenotípicas de la enfermedad de Fabry, aunque puede atribuirse en parte a la heterogeneidad de las mutaciones causales e incluso al tipo de sangre del paciente. Los pacientes con grupo sanguíneo B y AB (con residuos terminales de α- galactosil en sus membranas celulares) pueden tener un mayor sustrato corporal y tienen una enfermedad más agresiva comparada con pacientes con grupos sanguíneo A u O. Esta hipótesis no ha sido sistemáticamente estudiada, y se necesita más información de la historia natural de la patología que permita conocer los diversos factores que influyen en la expresión de la enfermedad de Fabry [8]. Histología de la Enfermedad de Fabry Estudios realizados empleando técnicas de microscopia de luz y electrónica para evaluar los cambios histológicos en tejidos de ratones knockout para α-Gal-A, mostraron que las células con mayor alteraciones son los macrófagos y las células derivadas de macrófagos del hígado y la piel, y las células musculares lisas que rodean a los vasos sanguíneos y el corazón [14]. En un estudio realizado en un paciente con enfermedad de Fabry, la mayor concentración de globotriaocilceramida (Gb3) se reportó primero en los riñones, seguido del corazón y el hígado; y esta fue 35 veces mayor en el riñón que en el hígado [16]; contrariamente cuando se hizo la misma observación en ratones knockout Gb3 fue similar en ambos tejidos. Otro aspecto interesante de resaltar de este estudio con ratones knockout son los obtenidos con el transplante de médula ósea, en donde en un número de órganos, la concentración de Gb3 fue significativamente menor en los ratones transplantados. El transplante alogénico de médula ósea, como fuente de enzima normal, ha indicado ser beneficioso en estudios humanos con otras enfermedades lisosomales, pero nunca ha sido utilizado en la enfermedad de Fabry [14,16]. En ratones knockout tratados con transplante de médula ósea, la actividad enzimática de α-Gal-A en el hígado fue de 10% con respecto a los niveles normales, lo que indica que la acumulación de Gb3 fue completamente depurada de éste órgano. En experimentos con cultivos de fibroblastos, bajos niveles de la actividad de α-Gal-A (menor de 5%) fueron capaces de normalizar el metabolismo del sustrato [14]. En las células cardíacas, los depósitos de Gb3 provocan una hipertrofia progresiva, y no es un fenómeno específico, ya que se observa en otras enfermedades de depósito lisosomal. Los depósitos observados en las células endoteliales están asociados con hipertrofia de la membrana basal del capilar y se han observado con o sin ningún grado leve de depósitos, lesiones ni en los sinusoides de la corteza de la glándula adrenal ni en el hígado. Los glomérulos renales muestran hialinización focal que se inicia en la región mesangial y las células tubulares proximales están libres de depósitos [17]. En los podocitos y en las células del epitelio tubular distal es donde se han observado la mayor cantidad de depósitos intracitoplasmáticos. La observación de los depósitos con el microscopio electrónico muestra inclusiones osmofílicas típicas en el citoplasma de todos los tipos de células renales y se observa la típica imagen de “piel de cebolla” o “apariencia de cebra”. Estos hallazgos patológicos son evidentes en pacientes homocigotos o heterocigotos, y ocurren antes de que se desarrolle la enfermedad renal clínica [18]. Las paredes de las arterias coronarias, renales e intrarrenales muestran signos de arteriopatía, con un importante compromiso de las células del músculo liso vascular. La arteriopatía se inicia con los depósitos, seguidos por la degeneración celular, depósito en la matriz extracelular y calcificación confinada a la pared vascular. Ocasionalmente, las células musculares lisas muestran necrosis, con dispersión de depósitos lipídicos dentro del citoplasma. Se han observado fibroblastos con grado variable de depósitos a nivel de la capa íntima de los vasos y de la válvula mitral, y esto está asociado con la presencia de pérdida de células y necrosis. Igualmente se han observado grandes depósitos a nivel de las células de Leyding y en epitelio del epidídimo [17]. La enfermedad de Fabry, junto con la enfermedad de Batten-Kufs´ (lipofuscinosis ceroide) y la manosidosis, forman un grupo de enfermedades de depósito lisosomal con manifestación asintomática en el músculo esquelético (Fig. 5), a diferencia de lo que ocurre en la Enfermedad de Pompe, que es otra enfermedad de depósito lisosomal, por deficiencia de la α-1,4 glucosidasa; pero con compromiso sintomático del músculo esquelético [19,20]. Aspectos Clínicos de la Enfermedad de Fabry La incidencia de la enfermedad de Fabry no se conoce de forma exacta pero oscila entre 1:40000 a 1:100000 sin diferencia entre razas [11]. Para el año de 1999, en los Estados Unidos, se estimaba la existencia de 5000 pacientes con esta enfermedad [16]. Según Pastores y col (2002) [8], esta enfermedad es un desorden huérfano pan-étnico con una incidencia de 1:117000 nacidos vivos. Sin embargo, dicha incidencia podría estar subestimada debido a que algunos pacientes con anormalidades cardíacas presentan una actividad enzimática residual y tales anormalidades no son extendidas a la enfermedad lisosomal. En otro estudio realizado por Kai-Chung Tse (2003) la evaluación de los pacientes con cardiopatía reveló en un 3.9% una deficiencia subyacente de la enzima α-galactosidasa A. La patología clínica de la enfermedad de Fabry depende del órgano o los órganos afectados [21]. A.-Enfermedad de Fabry Clásica. El fenotipo de la enfermedad de Fabry clásica en los pacientes de sexo masculino, con baja o casi ninguna actividad enzimática de α-Gal-A, está caracterizado por acroparestesias, angioqueratomas, hipohidrosis, y opacidad corneal y lenticular, desde la niñez o la adolescencia. A medida que la enfermedad avanza, la acumulación progresiva de Gb3 en los lisosomas del endotelio vascular, conduce a la manifestación de una insuficiencia renal, enfermedad vascular cerebral y cardíaca, y lo más seguro a una muerte prematura. En contraste, las mujeres portadoras del fenotipo Clásico presentan compromiso clínico muy variado que va desde ser asintomáticas o presentar manifestaciones moderadas, hasta llegar a tener la enfermedad, en un estadio de gravedad al igual que en los varones afectados [22]. En la siguiente sección se revisarán las características clínicas de los sistemas afectados con mayor frecuencia en la enfermedad de Fabry. A.1.-Compromiso Dérmico Se observan angioqueratomas distribuidos principalmente a nivel de hipogastrio y la zona del cinturón, genitales externos, palmas y plantas. El angioqueratoma comprende un hemangioma cavernoso dérmico con engrosamiento de la epidermis de revestimiento e hiperqueratósis en la misma (Fig. 6). Clínicamente se presenta como nódulos de color azul rojo ligeramente elevados, que raramente tienen más de 1 cm de diámetro [23]. Existen seis enfermedades de depósito lisosomal que producen angioqueratomas corporis diffusum. Los angioqueratomas más conocidos se advierten en los pacientes con enfermedad de Fabry, seguidos por los observados en la fucosidosis y galactosialidosis; además se han reportado en las enfermedades de Kansaki, beta-manosidosis y aspartylglucosaminuria [24]. Como parte de la investigación de la patogénesis de la hipohidrosis observada en la enfermedad de Fabry, Lao y col. (1998) realizaron en glándulas sudoríparas de un paciente con enfermedad de Fabry un estudio de microscopía electrónica, en el cual encontraron numerosas inclusiones citoplasmáticas en las glándulas, con un patrón laminar variable, en donde predominaron grandes inclusiones vacuolares en las células claras de la cola secretora; además en los conductos se observaron menos vacuolas, y las células basales del conducto mostraron figuras tipo mórula. Se observaron inclusiones laminadas en los axones amielínicos que inervan las glándulas. Los vasos sanguíneos que rodean a las glándulas exocrinas se mostraban con zonas muy estrechas debido a la presencia de células endoteliales hinchadas con inclusiones densas. La presencia de los depósitos intracitoplasmáticos podrían ser los responsables de la reducción en la capacidad de sudar, que muestran estos pacientes. Grupos de estas angiectasias cuyo color oscila entre rojo y azul oscuro, se encuentran entre el ombligo y el muslo de forma típica (“distribución en traje de baño”). Las lesiones no blanquean y con la edad se hacen más numerosas y más grandes. Su tamaño oscila desde pequeños puntos hasta varios milímetros (Tomado de Desnick RJ., 1979) [26] A.2.-Compromiso ocular Son comunes las lesiones vasculares de la conjuntiva y la retina; se observan opacidades cornéales, que comienzan con un patrón difuso y progresan hasta adquirir un patrón en espiral o radiado (Fig. 7); este último hallazgo, en los pacientes hemicigotos con la forma clásica de la enfermedad se observa casi invariablemente y en una alta proporción en las mujeres heterocigotas. Además, en los pacientes afectados por la enfermedad de Fabry se han observado dos tipos de opacidades lenticulares. Los depósitos color crema en la cápsula anterior del cristalino (algunas veces distribuidos en forma de hélice) son menos comunes que los depósitos granulares radiados, de color blanco, observados en la parte posterior del cristalino (conocidos como catarata de Fabry). Las cataratas posteriores de Fabry se encuentran en un 37% de los hemicigotos y en un 47% de los heterocigotos [27]. A.3.-Compromiso renal En los hombres con Enfermedad de Fabry durante la tercera década de su vida, se desarrolla una azotemia y proteinuria, no obstante antes de los 20 años de edad pueden ser detectados depósitos renales, excreción renal de Gb3, y la disfunción tubular. En pacientes masculinos de más de 50 años de edad la proteinuria es casi universal, y la progresión hacia la insuficiencia renal es tan alta como la observada en los pacientes diabéticos. Antes de que fuese necesario llevar a cabo los procedimientos que conllevan a la diálisis, los hombres afectados por la Enfermedad de Fabry Clásico, morían antes de los 50 años de edad [28]. La gráfica A muestra el daño típico de la conjuntiva, en done se observan los vasos similares a unas salchichas y marcadamente dilatados. La gráfica B muestra la opacidad corneal la cual es visible únicamente mediante microscopía con lámpara de hendidura. Esto no afecta la visión pero es un indicador útil, estando presente en los hemicigotos y en un 70-90% de los heterocigotos (Tomado de Desnick RJ, 1979) [26] Compromiso cardíaco En los pacientes con enfermedad de Fabry se ha descrito hipertrofia del ventrículo izquierdo y un aumento de la presión pulmonar sistólica, la cual se correlaciona íntimamente con la edad; igualmente se ha observado la presencia de lesiones valvulares tanto aórtica como mitral, así como anormalidades electrocardiográficas del segmento ST y falla en la conducción. Se puede presentar en ambos sexos una insuficiencia cardiaca, arritmias y cardiopatía isquémica, y los pacientes pueden desarrollar arritmias cardíacas complejas repetitivas, antes del desarrollo de la hipertrofia ventricular izquierda [28]. La hipertrofia ventricular se debe a depósitos en los miocitos cardíacos de glucoesfingolípidos y a un incremento absoluto en la masa muscular cardiaca [21]. La enfermedad de Fabry ha sido considerada causa de hipertrofia ventricular izquierda en el 6% de los hombres que presentan cardiomiopatía hipertrófica de aparición tardía, y es la causa de miocardiopatía hipertrófica de aparición tardía en el 12% de las mujeres, siendo ellas posibles candidatas a recibir el tratamiento de reemplazo enzimático [29]. A.5.-Compromiso neurológico La expresión clínica de la enfermedad a nivel del sistema nervioso puede presentarse en forma temprana en el curso de la misma, y se caracteriza por ataques isquémicos transitorios, lesiones de la sustancia blanca, hemiparesia, vértigo, diplopia, disartria, nistagmus, tinnitus, hemiataxia, amnesia e hipoacusia [28]. Estas manifestaciones son debidas a ectasia de grandes vasos, enfermedad oclusiva de los grandes y pequeños vasos. La diátesis vascular tiene una distribución vertebrobasilar. Las acroparestesias, son dolores neuropáticos que afectan a las terminaciones nerviosas distales [21]. Además, se ha considerado que la hipohidrosis afecta al sistema nervioso autónomo a nivel de las glándulas sudoríparas. Mac Dermon y col. (2001) estudiaron 98 hombres con enfermedad de Fabry y observaron el dolor neuropático en 93 de ellos (77%), con un promedio de soporte del dolor en un valor de 5 (escala de 0 a 10). Adicionalmente, encontraron complicaciones cerebrovasculares en el 24.2% de los individuos estudiados. La hipoacusia sensorioneuronal para sonidos de alta frecuencia se presentó en el 78% de las audiometrías realizadas. Igualmente, Germain DP, (2000) [29] observó que la hipoacusia progresiva es un hallazgo frecuente en la enfermedad de Fabry; por otra parte también investigó la función coclear en 22 varones homocigotos con enfermedad de Fabry Clásico (con edades entre 19 y 64 años), encontrando alteraciones auditivas en 12 de los pacientes (54.5%), de los cuales en 5 de ellos fue la pérdida progresiva de la audición y en los otros 7 una sordera aguda. En 7 de los 10 pacientes restantes se observó hipoacusia para los tonos de alta frecuencia, sin compromiso clínico, a pesar de ser jóvenes al momento del examen. La incidencia de hipoacusia parece estar significativamente aumentada en pacientes con daño renal (p<0,01), o daño cardíaco (p<0,01), mientras que no encontraron correlación con la hipertrofia ventricular izquierda. Tratamiento clínico de la Enfermedad de Fabry Durante muchos años el tratamiento de la enfermedad de Fabry fue de tipo paliativo de acuerdo a los síntomas del paciente, a saber: hidratación para mejorar la sensación de sofocamiento, analgésicos en caso de dolores musculares o viscerales; anticonvulsivantes como la carbamacepina para aliviar las acroparestesias, y la diálisis en caso de falla renal terminal. Sin embargo, durante todo el siglo XX, se investigó en la búsqueda de una terapia específica para esta enfermedad, orientada a la obtención de la enzima específica que pudiera revertir el daño causado por la deficiencia de la enzima antóloga. IV.1.-Terapia Enzimatica Estudios iniciales pilotos sobre la terapia de reemplazo enzimático en hombres con enfermedad de Fabry Clásico, consistieron en la administración endovenosa, tanto de una dosis única de plasma fresco que contenía α-Gal-A activa como una dosis única de enzima placentaria parcialmente purificada [30,31]. Estos estudios demostraron que la enzima obtenida de plasma o de placenta administrada por vía endovenosa, podía disminuir el nivel de Gb3 acumulado en el plasma. En 1973, Kornfeld y col. [32], inyectaron la enzima obtenida de placenta humana, y en 1979, Desnick y col. [26] suministraron 6 dosis de preparación enzimática obtenida de plasma o de bazo. En ambos estudios se evaluaron a 6 pacientes y se encontró que la concentración de Gb3 circulante se reducía transitoriamente y se regresaba a valores de pre-infusion en 48 horas. Estas investigaciones no determinaron los efectos de α-Gal-A en los niveles tisulares de Gb3 [33]. En un estudio llevado a cabo por Desnick y col. (1979) [26] en donde a dos hermanos que padecían la enfermedad le administraron 6 dosis de α-GAL-A purificada de tejido esplénico o plasma, en un período de 3 meses; encontraron que la enzima de origen esplénico se depurada rápidamente de la circulación (tiempo de vida media 10 minutos), diminuyendo transitoriamente de la circulación la concentración de Gb3; mientras que la enzima derivada del plasma, más altamente sialilada, tuvo una depuración más lenta (tiempo de vida media de 70 minutos) y mayor disminución de Gb3 circulante. Posteriormente, en 1980 Desnick reportó una vez más, que dos dosis de la enzima derivada plasma administrado los días 1 y 3, reducían los niveles casi normales, el nivel de sustrato plasmático. Estos estudios demostraron la viabilidad de la terapia de reemplazo enzimático para la enfermedad de Fabry, el mayor obstáculo para enfrentar el problema era la dificultad para a producir suficiente enzima purificada [34,35]. La viabilidad del tratamiento de la enfermedad de Fabry con la Terapia de Reemplazo Enzimático (TRE) con α-Gal-A, se basa en el hecho de que las enzimas lisosomales que escapan al sistema de transporte del receptor del aparato Golgi al compartimiento prelisosomal, son secretadas por las células y con frecuencia son recaptadas por los receptores de manosa-6-fosfato de la superficie celular, que regresan la enzima al lisosoma por endocitosis. El estudio de Schifmann y col (2000) [33] utilizando TRE con α-Gal-A, en pacientes con enfermedad de Fabry, demostró que ésta enzima obtenida de fibroblastos humanos es segura y bien tolerada a cualquier dosis. Histoquímica del hígado mostró que la infusión de α-Gal-A está distribuida en varios tipos de células. La mayor cantidad de la enzima, se detectó en las células de Kupffer y en las células endoteliales sinusoidales; pero también se detectaron cantidades significativas de la enzima en los hepatocitos. La enzima α-Gal-A, una vez unida a su receptor de manosa-6-fosfato, es internalizada en la célula que contiene el depósito de Gb-3. En todas las células renales de pacientes adultos con enfermedad de Fabry, se ha reportado abundante cantidad de Gb-3; pero especialmente en el epitelio glomerular y en las células tubulares renales. Es por ello que en el sedimento urinario, puede medirse la presencia de glucoesfingolípidos acumulados en las células tubulares descamadas. En el día 28 después de la administración de la enzima en la dosis indicada, se observó en el sedimento urinario un descenso significativo en la excreción de Gb-3. La reducción tardía de Gb-3 podría reflejar el lento intercambio de las células tubulares, y también señalaba una entrega o un aporte efectivo y eficiente de la enzima a las células epiteliales del túbulo renal; además, estos resultados sugieren que la enzima es activa por un período de tiempo significativo y provee una correlación funcional para la observación de una vida media hepática prolongada. Estos resultados demostraron que las infusiones únicas de α-Gal-A obtenida de fibroblastos humanos transfectados son seguros y bioquímicamente activos en pacientes con enfermedad de Fabry. La actividad biológica de la enzima está determinada principalmente, por el tiempo de permanencia de la enzima activa, dentro del lisosoma de varios tejidos. La vida media corta y larga en plasma en los tejidos, respectivamente, sugiere que la enzima puede administrarse semanal o quincenalmente [33]. Se han obtenido 4 isoformas de α-Gal-A recombinante: AGA-1, AGA-2, AGA-3 y AGA 4, las cuales varían en el número de residuos de ácido siálico y manosa-6-fosfato, las cuales tienen una farmacocinética y una distribución tisular similar. Aunque las formas glicosiladas más sialiladas (AGA-1 y AGA-3) permanecen por más tiempo en la circulación, la biodistribución de las 4 isoformas fue similar; ya que aproximadamente el 95% de cada una de ellas era recuperada en el hígado y el resto (5%) en el bazo y el riñón. Probablemente, la mayor captación hepática se debe a la presencia de los receptores de manosa en el sistema reticuloendotelial, y de los receptores de manosa-6-fosfato lisosomal intrahepático. El Gb3 acumulado en el hígado se depuraba rápidamente, y por más de 4 semanas permanecía indetectable, mientras que las concentraciones de Gb3 a las 2 y 3 semanas post-inyección en el bazo y el corazón disminuyeron notablemente respectivamente, posterior a lo cual comenzaba a reacumularse en dichos órganos. Estos hallazgos sugieren que la administración quincenal de α-Gal-A puede degradar el Gb3 acumulado y prevenir su reacumulación. Se observó un descenso pronunciado del depósito lisosomal de glucolípidos en los pericitos del músculo liso vascular, en fibroblastos, histiocitos del corazón, en las células de Kupffer y en los túbulos renales. La observación de numerosas vacuolas translúcidas limitando el sinusoide hepático después de la administración de la enzima, sugiere que la enzima α-Gal-A es endocitada dentro de los endosomas, para su posterior distribución a los lisosomas, que contienen el sustrato depositado. Debido a que la enzima α-Gal-A no es activa a pH sanguíneo neutro, y que los glucoesfingolípidos circulantes son transportados por lipoproteínas plasmáticas sintetizadas en el hígado, se sugiere que después de la administración de la enzima, el descenso en los niveles plasmáticos de Gb3, es el resultado de la depuración del sustrato depositado en los tejidos [35]. Para las infusiones de r-hα-Gal-A, los datos del tiempo de concentración mostraron un perfil no lineal, dosis-dependiente, consistente con una depuración de la enzima circulante, a través de las vías saturables y no saturables. A los 60 minutos de una infusión de 0.3 mg/kg de peso, y a los 90 minutos de una infusión de 1 y 3 mg/kg de peso, la concentración plasmática promedio alcanzó el 80% del valor pico. Cuando las infusiones se completaron, las concentraciones promedio cayeron a la mitad del valor pico dentro de los 15, 20 y 45 minutos para concentraciones de 0.3 a 3 mg/kg de peso, respectivamente. Para todos los grupos de infusión, las concentraciones plasmáticas de Gb3 se redujeron de una forma dosis-dependiente. Antes del tratamiento, todos los pacientes tenían niveles plasmáticos de Gb3 elevados, en un rango entre 2 a 53.9 ng/µl (promedio de 17.1 + 12.8 ng/µl); el nivel normal de Gb3 es menor de 1.2 ng/ µL. A una dosis de 0.3 mg/kg de peso, los niveles plasmáticos de Gb3 en pacientes que reciben r-hα-Gal-A quincenal, tendieron a disminuir con cada administración, alcanzando un valor bajo en la quinta infusión. En contraste, en los pacientes que recibieron la enzima a la dosis de 3 mg/kg de peso, quincenalmente, los niveles de Gb3, depuraron totalmente después de la primera infusión, y permanecieron indetectable a los largo de todo el estudio. Dos de los tres pacientes que recibían la enzima recombinante a la dosis de 1 mg/kg de peso, quincenalmente, mostraron también una depuración de Gb3 plasmática después de la primera infusión, mientras que el tercer paciente tuvo una reducción de los niveles de Gb3 plasmático; pero nunca alcanzó niveles indetectables. Los pacientes que recibían la enzima a la dosis de 1 ó 3 mg/kg de manera interdiaria, mostraron niveles de Gb3 bajos a la cuarta dosis, sin embargo, fueron menores que los niveles observados en pacientes con un esquema quincenal [36,37]. Las variaciones en los niveles celulares de glucoesfingolípidos y en la respuesta tisular a la enzima recombinante, reflejan la síntesis endógena de glucolípidos y la tasa de recambio celular, mientras que la diferencia en la depuración es una función de la captación celular y lisosomal. En base a los estudios pre-clínicos en ratones con enfermedad de Fabry, se sabe que el hígado es el órgano que muestra los niveles más altos de captación enzimática; todos los regímenes de administración de la enzima reducen marcadamente los niveles de Gb3 en ese órgano, y en pacientes se ha observado la depuración hepática de Gb3 en un 84%, mediante la utilización de la metodología de ELISA, en las biopsias pre- y post-tratamiento. Las células endoteliales de los sinusoides hepáticos y las células de Kupffer fueron casi totalmente depuradas de los glucoesfingolípidos. [36]. A nivel renal, el contenido vascular total de Gb3 es un componente relativamente menor de la acumulación total de Gb3; además, en los podocitos las inclusiones no se modificaron, por lo tanto la mayor reducción de Gb3 proviene del túbulo, lo cual es el otro gran depósito de Gb3. La alta depuración de Gb3 tubular, junto con la depuración del endotelio vascular, sugieren el beneficio potencial de la función renal [36]. De todo lo que se ha planteado en esta revisión, se puede concluir que la Enfermedad de Fabry es una Endoteliopatía de origen enzimático, con compromiso multisistémico. Además es una Enfermedad de abordaje multidisciplinario, para la pesquisa, el diagnostico, tratamiento y pronóstico de la Enfermedad. Por último, actualmente se puede decir que la enzimoterapia es una realidad con resultados satisfactorios que mejoran la calidad de vida de los pacientes y sus familiares. Referencias
NOTA: Toda la información que se brinda en este artículo es de carácter investigativo y con fines académicos y de actualización para estudiantes y profesionales de la salud. En ningún caso es de carácter general ni sustituye el asesoramiento de un médico. Ante cualquier duda que pueda tener sobre su estado de salud, consulte con su médico o especialista. Copyright 2008 - Centro de Análisis de Imágenes Biomédicas Computarizadas CAIBCO, Instituto de Medicina Tropical – Facultad de Medicina, Universidad Central de Venezuela The following images related to this document are available:Photo images[va08020t1b.jpg] [va08020t1.jpg] [va08020t1a.jpg] [va08020f1.jpg] [va08020f3.jpg] [va08020f2.jpg] [va08020f7.jpg] [va08020f5.jpg] [va08020f4.jpg] [va08020t2.jpg] [va08020f6.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}