 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
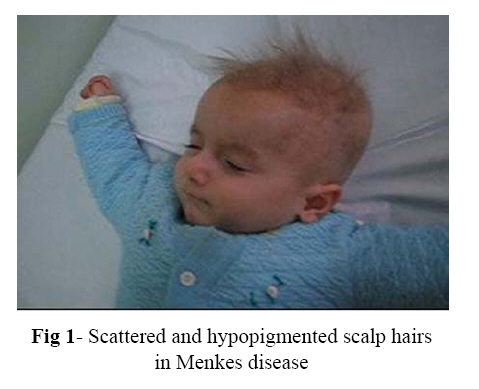
Iranian Journal of Pediatrics, Vol. 17, No.4, December 2007, pp. 388-392 Case Report Menkes Disease: Report of Two Cases Mohammad Barzegar*1, MD; Afshin Fayyazie1, MD; Bobollah Gasemie2, MD;Mohammad ali Mohajel Shoja3, MD 1Pediatric Neurologist, Department of Pediatrics, Tabriz University of Medical Sciences, IR Iran2Pathologist, Department of Pathology, Tabriz University of Medical Sciences, IR Iran 3General Physician, Tabriz University of Medical Sciences, IR Iran * Correspondence author; Address: Pediatric Department, Tabriz Children Hospital, Sheshgelan St, Tabriz, IR Iran. E-mail: mm_barzegar@yahoo.com Received: 14/05/07; Revised: 20/08/07; Accepted: 10/10/07 Code Number: pe07073 Abstract Introduction: Menkes
disease
is
a
rare
X-linked
recessive
disorder
of
copper
metabolism.
It
is
characterized
by
progressive
cerebral
degeneration
with
psychomotor
deterioration,
hypothermia,
seizures
and
characteristic
facial
appearance
with
hair
abnormalities. Key Words: Menkes disease, Copper metabolism, Epilepsy, Pili torti, Cerebral degeneration IntroductionMenkes disease (MD), also referred to as kinky hair disease, trichopoliodystrophy, and steely hair disease, is a rare X-linked recessive disorder of copper metabolism.[1,2] It is characterized by progressive cerebral degeneration with psycho-motor deterioration, seizures, and connective tissue alteration with hair abnormalities.[2,3] The most striking finding is the appearance of the scalp hair, being thin, coarse, brittle and colorless. Examination under microscope reveals a variety of abnormalities, most often pili torti (twisted hair), monilethrix (varying diameter of hair shafts) and trichorrhexis nodosa (fractures of the hair shaft at regular intervals)[4]. The clinical picture is caused by a defect in copper transporting ATPase (ATP7A), resulting in defects of key copper dependent enzymes, including lysyl oxydase, cytochrome c oxidase, dopamine β-hydroxylase, tyrosinase, and super oxide dismutase. Depigmentation of hair and skin pallor are due to tyrosinase deficiency, hypothermia is due to cytochrome c oxidase deficiency and lysyl oxidase deficiency causes tortuous arteries in brain, progressive vascular changes predispose to thrombosis and deficient blood supply to the developing brain[2,3,5]. Neuroimaging discloses atrophy and bilateral ischemic lesions in deep gray matter or in the cortical areas; the consequence of vascular infarctions.[7] Management of patients with MD is supportive, with an emphasis on anticonvulsant treatment and a trial of copper histidine therapy. Prognosis is poor with progressive neurological deterioration and eventual death within the first 3 years of life.[3] The clinical history and the appearance of the infant should suggest the diagnosis. Microscopic examination of the hair is very helpful even in a mild case. Low levels of serum copper and ceruloplasmin will usually confirm the diagnosis.[5] If doubt still exists, the diagnosis can be confirmed by demonstrating the intracellular accumulation of copper and decreased efflux of 64Cu from cultured fibroblasts.[6] Menkes disease is a rare disorder; its frequency has been estimated 1 in 114000-250000 live births.[8] We report on two cases of classic MD diagnosed in Tabriz Children's Hospital, a university- affiliated tertiary hospital in the East Azarbaijan province, the North West of Iran between years 2002 and 2006. Case(s) PresentationCase 1: A seven month-old male infant was brought to our out-patient clinic due to gradual-onset of hypotonia and seizures. The boy was born at 34 weeks of gestational age to healthy, non-consanguineous parents. He was the first child of the parents. His early development was age appropriate for 3 months, and then regressed. At 5 months of age myoclonic jerks were noted. His clinical examination at 7 months revealed cherubic appearance with depressed nasal bridge, and brittle, scattered and hypopigmented scalp hairs (Fig 1). He had no eye contact and no head control. Light microscopic examination of the scalp hair showed pili torti. The diagnosis of MD was confirmed on the basis of low serum copper (15 μg/dl; ref. range: 70-150 μg/dl) and low serum ceruloplasmin (58 mg/l; ref. range: 187-322, mg/l). There were no abnormalities in other standard blood analyses. The electroencephalo-graphy showed multifocal spikes and waves with poorly organized sleep features. Brain CT scan demonstrated cerebral atrophy and subdural effusion. Copper-histidine was prescribed. Despite anticonvulsant therapy with various drugs (phenobarbital, clonazepam, nitrazepam, vigabatrin) intractable seizures continued. At the end of the first year of life, neurological milestones such as head control, rollover response and laughing had not been achieved. Unfortunately, the patient died of a respiratory infection at the age of 14 months. Case 2: The five month-old male infant was referred to our hospital with regression of developmental milestones and seizures. He was born at term to healthy consanguineous parents. The pregnancy was uneventful. At birth the head circumference and body weight were 35 cm and 3.2 kg, respectively. Family history was remarkable for the death of two previous male siblings at the age of 1 month and 18 months. They had severe neurodevelopmental delay without a definite diagnosis. The patient had a history of 8 days hospital admission on third day of life with poor feeding, hypothermia and hyperbilirubinemia (total bilirubin was 16 mg/dl). His early development was age appropriate for 4 months, at 3 months of age he had good head control and laughing. At 4 months of age tonic and myoclonic seizures were noted. On clinical examination at 5 months, the most striking finding was the appearance of the scalp hair. It was colorless, thin, brittle and kinky. Although eye contact was noted, he had poor head control and no rollover response. Brain CT scan showed cerebral atrophy and subdural effusion. An electroencephalography revealed frequent multifocal epileptiform discharges with disorganized background. With high suspicion of MD, serum copper and ceruloplasmin were determined; with 3 μg/dl (ref. 70-155) and 15 mg/l (ref. 187-320 mg/l) respectively both were below normal levels. Light microscopic examination of the hair showed pili torti (twisted hair shafts) and trichorrhexis nodosa. There were no metaphyseal changes of long bones on X-rays. There were also no abnormalities in other standard blood analyses. The diagnosis of MD was made. Phenobarbital and nitrazepam were partially effective. Poor weight gain, seizures and neurologic deterioration were evident in a visit at 1 year of age. At 17 months of age he showed severe global developmental delay and failure to thrive. He has had two short hospital admissions for chest infection and diarrhea. DiscussionThe clinical features and inheritance of MD were first described in 1962.[9] Ten years later the underlying biochemical defect in copper metabolism was discovered.[10] The clinical spectrum of MD encompasses several distinct variants. The neonatal form is characterized by multiple fractures and extensive vascular disease with early death[11]. Infants with classic MD typically appear healthy until 2 to 3 months of age. Premature delivery is very frequent, as are neonatal hypothermia and hyperbilirubinemia. Hypothermia may also occur in older infants. Neonatal symptoms may resolve, and the babies may seem normal during next 2 or 3 months. At 3 months of age they start to demonstrate developmental delay, hypotonia, intractable seizures and failure to thrive. Cerebral degeneration then dominates the clinical picture.[1,3,5] Children often have a cherubic appearance with sparse, course, short, twisted, and lightly pigmented hair. Individuals with the mild variant are developmentally delayed with cerebellar ataxia, dysarthria and pili torti, and no seizures. The occipital horn syndrome is considered a MD variant. The skeletal dysplasia, soft bruisable skin, hyper-extensible joints, diarrhea, and occipital exostosis characterize it.[12] The typical history and clinical features in our patients were suggestive of classical MD. The cherubic facial appearance with a depressed nasal bridge in both of them was similar to reported cases in the past.[1-3,5,6,9,13] Hair abormalities are the most striking signs in this syndrome. Our patients showed brittle, scattered and hypopigmented scalp hairs; under microscope hair was shown to be twisted longitudinally with narrowing at intervals along-with many erosions on the hair shaft. The hair was fragile and fractured easily, resulting in apparent generalized alopecia. Several hair shaft abnormalities have been documented, with pili torti being the most common, also trichorrehexis nodosa, trichoclasis, and trichoptilosis have been reported.[4,14] The scalp hair may appear normal at birth, but at approximately three months of age the hair on the scalp and eyebrows becomes kinky, coarse, and lightens in color.[1,3,5] Non–skin manifestations in our patients were delayed developmental milestones and intractable seizures similar to other reports.[1,3,5,6,9] Epilepsy is a frequent and early feature in MD, it was reported in our cases. Myoclonous is the usual seizure type; other types of seizures, including multifocal seizure and tonic spasms are also reported. Seizures are usually resistant to antiepileptic drugs. The pathophysiologic mechanisms of epilepsy in MD remain unknown, but they are likely related to copper deficiency. It results in an impairment of lysyl oxidase, considered as the primary cause of the abnormal intracranial vessel structures.[15] Our patients had low serum copper and ceruloplasmin levels which correlated with the clinical findings, these levels are usually low but interpretation may be difficult in the first few months of life[5]. In the past, the final diagnosis was made by cultured skin fibroblasts and lymphoblasts, which showed impaired metabolism of copper.[6] Today this method is being replaced by molecular genetic analysis, available in certain laboratories, to confirm the diagnosis for carrier testing or for prenatal diagnosis.[16] Our diagnosis was made on the basis of clinical and laboratory findings. Our cases fulfilled the following clinical and biologic diagnostic features of the classical MD: Progressive neurologic disease with marked hypotonia and neurologic regression, feeding difficulties with failure to thrive, cherubic appearance with a depressed nasal bridge, brittle, scattered and hypopigmented scalp hairs, pathognomonic hair abnormalities consistent with pili torti, and low plasma copper and serum ceruloplasmin levels. Copper histidine is the most effective treatment for MD, and if admiminstered soon after birth, neurological development can be maintained[17]. By contrast, there are few neurological benefits when Cu-histidine treatment is initiated after 2 months of age.[18] Therefore it would be important to have it easily available in drug market. The early diagnosis is also mainly required for the genetic counseling. The pattern of inheritance is X-linked with a recurrence risk of 50% for the affected sons and 50% of daughters will be carriers. Unfortunately, in our second case genetic analysis was not available. Prenatal diagnosis of MD can be performed by gene analysis and/or measurement of the copper concentration in culture of amniotic liquid cells and chorionic villi cells.[16] ConclusionWe believe that MD is an under-diagnosed entity in the developing countries, so being familiar with its manifestation and maintaining high index of suspicion are necessary for early diagnosis. The diagnosis can be made with great confidence by the typical clinical history and the appearance of the infant once one or two cases have been seen. References
Copyright 2007 - TUMS PUBLICATIONS The following images related to this document are available:Photo images[pe07073f1.jpg] |
| |||||||||

{kind=link}