 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Tropical Journal of Pharmaceutical Research, Vol. 4, No. 1, June 2005, pp. 369-375 Research Article Formulation and in vitro Evaluation of Eudragit® Microspheres of Stavudine Sunit Kumar Sahoo1, Abdul Arif Mallick1, BB Barik1 and Prakash Ch Senapati2 1University Department of Pharmaceutical
Sciences, Utkal University, Bhubaneswar, Orissa –751004, India, 2Torrent Pharmaceuticals, Baddi Code Number: pr05007 Abstract Purpose: The aim of this study was to formulate
and evaluate microencapsulated controlled release preparations of a highly
water-soluble drug, stavudine, using Copolymers synthesized from acrylic
and methacrylic acid esters (Eudragit RS 100 and RL 100) as the retardant
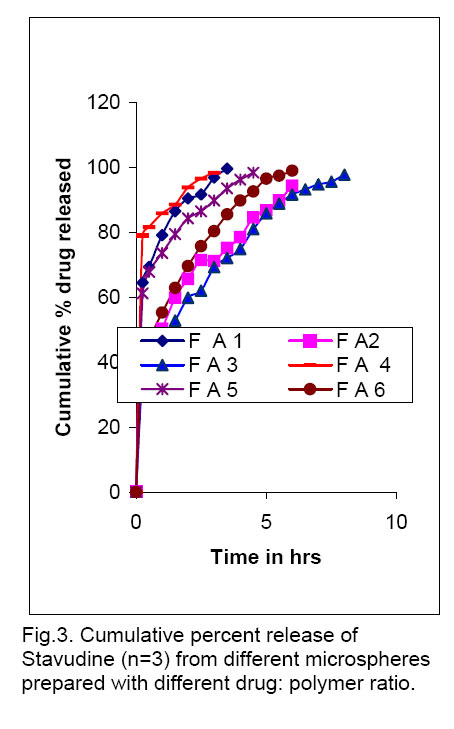
material. . KEYWORDS: stavudine, Eudragit, microspheres, controlled release, polymethacrylate. INTRODUCTION The population of patient with chronic disease or complications of other disease has recently been increasing. These situations necessitate taking drug for a long period and / or multiple medicines simultaneously, which can lead to increase in non-compliance. The problem would be worse for drugs with short biological half-life. One method to solve such problems is to find a dosage form capable of releasing the drug gradually. Microencapsulation has been used as one of the methods to deliver drugs in a controlled manner1. Stavudine (D4T, thymidine) is the FDA-approved drug for clinical use for the treatment of HIV infection, AIDS and AIDS-related conditions either alone or in combination with other antiviral agents. Stavudine is typically administered orally as a capsule and an oral solution. The virustatic drug has a very short half-life (1.30 h). However patients receiving stavudine develop neuropathy and lactic acidosis. The side effects of stavudine are dose-dependent and a reduction of the total administered dose reduces the severity of the toxicity2. Microencapsulated techniques have mostly been used for lipophilic drugs since hydrophilic drugs showed low loading efficiency. The objective of the present investigation was to prepare the controlled release, microspheres of stavudine by improving biological half-life and entrapment efficiency. One method of ensuring high entrapment efficiency of water-soluble active ingredients is to use a hydrophobic processing medium into which the hydrophilic drug molecule is unlikely to migrate out3. In this present study, stavudine microspheres were prepared by solvent evaporation technique using Eudragit RS 100 and RL 100 as a matrix polymer4. Liquid paraffin and acetone system were used for the preparation of microspheres. Magnesium stearate was used as a droplet stabilizer to prevent droplet coalescence in the oil medium and n-hexane was added as a non-solvent to the processing medium to solidify the microspheres5. The effect of various processing and formulation factors such as drug to polymer ratio, stirring speed and surfactant concentration on the mean particle size of microspheres was investigated. The prepared spherical microspheres were evaluated for micromeritic properties and drug content, and also by FTIR, DSC, X-RD, SEM as well as for in vitro drug release studies6. MATERIALS AND METHODS Materials Stavudine was obtained as a gift from Hetero Labs Ltd. (Hyderabad, India). Eudragit RS 100 and RL 100 was obtained from Röhm Pharma, GmbH, Darmstadt, Germany. All other reagents and solvents used were of pharmaceutical or analytical grade. Methods Stavudine microspheres were prepared by solvent evaporation techniques. Different amounts of Eudragit RS or Eudragit RS: RL combination was dissolved in 25 ml acetone separately by using a magnetic stirrer (Remi Equipments, model 2MIH). Pure stavudine (1 g previously dissolved in 10 ml methanol) and magnesium stearate [100 mg] were dispersed in the polymer solution. The resulting dispersion was then poured into 1000 ml beaker, containing the mixture of 270 ml liquid paraffin light and 30 ml n-hexane, while stirring. A mechanical stirrer with a blade [4 cm diameter] (Remi Motors, Model No.RO-123R, Mumbai) was used. Stirring (at 500-700 rpm) was continued for 3 h, until acetone evaporated completely. After evaporation of acetone, the microsphere formed were filtered using Whatman no.1 filter paper. The residue was washed with 4-5 times in 50 ml petroleum ether (400 C-600 C) each. Microspheres were dried at room temperature for 24 h. Formulations containing 1, 2 and 3 g of Eudragit RS only were assigned batch code as: FA1, FA2 and FA3 respectively and formulation with Eudragit RS: RL combinations as, 0.8:0.2,1.8:0.2 and 2.8:0.2 g were assigned batch code: FA4, FA5 and FA6 respectively. All batches were prepared in triplicate.
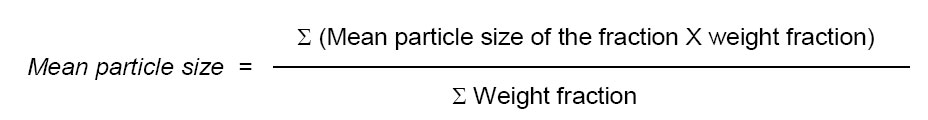
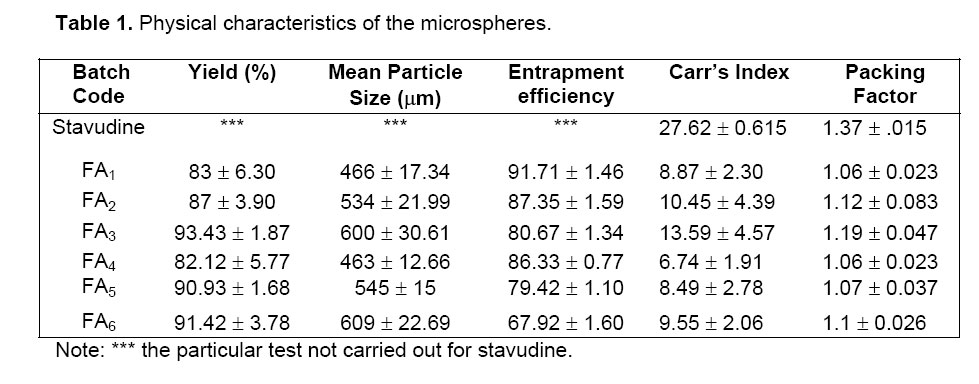
Microspheres dried at room temperature were then weighed and the yield of microspheres preparation was calculated using the following formula6: Measurement of Micromeritic properties of microspheres The flow properties of prepared microspheres were investigated by measuring the bulk density, tapped density, Carr’s index and packing factor. The bulk and tapped densities were measured in a 10 ml graduated measuring cylinder. The sample contained in the measuring cylinder was tapped mechanically by means of constant velocity rotating cam. The initial bulk volume and final tapped volume were noted from which, their respective densities were calculated. Each experiment was carried out in triplicate.
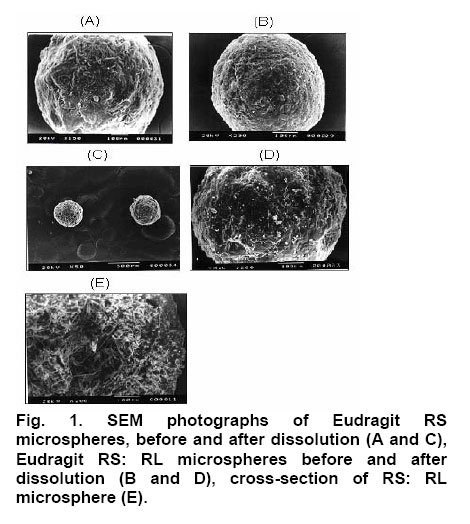
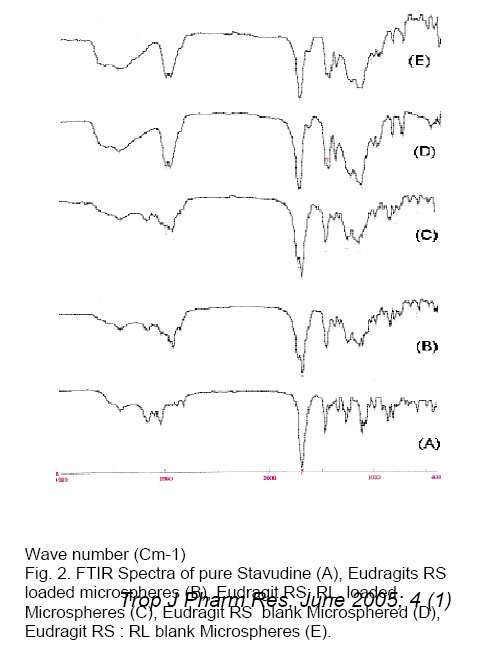
Drug entrapment efficiency About 50 mg of accurately weighed drug-loaded microspheres were added to 50 ml of phosphate buffer, pH 6.8. The resulting mixture was shaken in a mechanical shaker for 24 h. The solution was filtered with a 0.45 µm pore size filter and 1 ml of this solution was appropriately diluted to 25 ml using phosphate buffer, pH 6.8, and analyzed spectophotometrically at 266 nm using Systronic 2101 UV-Visible double beam Spectro-photometer. Scanning Electron microscopy (SEM) Scanning electron microscope (JEOL JSM -5200) was used to characterize the Shape and surface topography of the microspheres8. Prior to examination, samples were gold sputter-coated to render them electrically conductive. Fourier Transform Infrared Spectroscopy (FTIR) Drug-polymer interactions were studied by FTIR spectroscopy. The spectra were recorded for pure drug and drug-loaded microspheres using FTIR JASIO (Model No. 410). Samples were prepared in KBr disks (2 mg sample in 200 mg KBr). The scanning range was 400-4000 cm -1 and the resolution was 2 cm -1. Differential Scanning Calorimetry (DSC) The DSC analysis of pure drug and drug-loaded microspheres were carried out using a Diamond DSC (Perkin Elmer, USA) to evaluate any possible drug-polymer interaction9. The analysis was performed at a rate 5.000 C min -1 from 500 C to 2000 C temperature range under nitrogen flow of 25 ml min -1. X-ray powder Difftactometry [X-RD] X-ray powder diffractometry was carried out to investigate the effect of microencapsulation process on crystallinility of drug. Powder X-RD patterns were recorded on Rigaku, Japan (Model-MenifleX) using Ni-filtered, Cuk α radiation, a voltage of 30 kV and a current of 25 mA. The scanning rate employed was 20 min -1, over the 40 to 400 diffraction angle (2θ) range. The X-RD patterns of drug powder and drug-loaded microspheres were recorded. Drug release studies The in vitro release studies of drug-loaded microspheres were carried out at 370 C and 100 rpm using phosphate buffer pH 6.8 (500 ml) in a USP dissolution apparatus (LABINDIA, DISSO-2000, Mumbai, India) under sink conditions. Accurately weighed samples of microspheres (containing approx. 50 mg of drug, size fraction 250 µm) were added to dissolution medium and at preset time intervals 2 ml aliquots were withdrawn and replaced by an equal volume of fresh dissolution medium. After suitable dilution, the samples were analyzed spectophotometrically at 266 nm. The concentration of stavudine in test samples was corrected and calculated using a regression equation of the calibration curve. Release Kinetics Data obtained from in vitro release studies were fitted to various kinetics equations10 to find out the mechanism of drug release from microspheres. The kinetics models used were zero order, first order, and Higuchi models. The rate constants were also calculated for the respective models. RESULTS & DISCUSSION Effect of various processing and formulation parameters on mean particle size It was observed that when the speed of stirrer was below 500 rpm, there was no formation of spherical microspheres. This could be due inadequate agitation to disperse the inner phase in the total mass. Therefore, particles were found to settle at the bottom of vessel. At stirrer speeds of 700-1000 rpm, the resulting high turbulence, caused frothing and adhesion to the container wall. Therefore, the mean particle size of microshperes decreased. The desired spherical microspheres were obtained at stirring speeds of 500-700 rpm. When 50 mg magnesium stearate was incorporated, microspheres were not formed because the low magnesium content failed to prevent droplet coalescence in the oil medium; as a result mean particle size was increased. The mean particle size decreased with increasing amount of magnesium stearate (150 mg). This is probably a consequence of stabilization of the oil droplets with magnesium stearate. Spherical microspheres were formed when the magnesium stearate content was maintained at 100 mg. When the drug: polymer ratio was 1: 1, there was formation of microspheres with small and irregular size, and as the polymer concentration was increased, solution viscosity also increased, resulting in large particles. Thus, mean particle size also increased. Flow properties of Microspheres The flow properties are expressed in terms of Carr’s Index (see Table 1.). The Carr’s index for all formulations was less than 20, which indicates excellent flow compared to the original drug crystals. Also the microspheres were found to exhibit higher packing properties than the original drug crystals. The improvement in flow properties suggests that the microspheres can be easily handled during processing. Scanning Electron Microscopy (SEM) SEM study shows that particles were spherical. The surface of the drug-loaded microspheres manifested the presence of drug particles (see Fig. 1). The spherical nature and size of the microspheres did not change after the dissolution tests, but the number of pores increased. When a cross section of the microspheres were viewed it showed a spongy appearance. Fourier Infrared Spectroscopy (FTIR) As shown in Fig. 2, there was no significant difference in the IR spectra of pure stavudine and drug-loaded microspheres (Both Eudragit RS and RS: RL loaded microspheres). The characteristic OH stretching, NH stretching of secondary amine, C-H stretching and C=O stretching of pure drug was unchanged in case of microspheres. The results suggest drug stability during the encapsulation process. This was further supported by the DSC results that follow. Differential Scanning Calorimetry (DSC) The drug may have been dispersed in crystalline or amorphous form or dissolved in the polymeric matrix during formation of the of microspheres. Any abrupt or drastic change in the thermal behavior of either the drug or polymer may indicate a possible drug-polymer interaction11, 12. The thermo-gram of pure stavudine shows an endotherm at 1660 C., which correspond to its melting point (1610 C to 1670 C). This endotherm was also observed for Eudragit RS100 microspheres at 1600 C but it was less sharp and this suggests that there is a significant reduction in drug crystallinity in the polymer matrix. X-ray Powder Diffractometry (X-RPD) The X-ray powder diffraction patterns of pure drug, RS and RS: RL microspheres containing stavudine reveals that the intensity of the peaks for the pure drug was sharp. But when it was incorporated into the polymer matrix, the drug peaks showed a loss of sharpness due probably decreased crystallinity of the drug. Drug release behavior The pure stavudine showed a fast release as 98 % was released within 7 min. When it was encapsulated, sustained release up to 8 h was observed. For FA3, It was found that the release rate of drug from size fractions of 355 and 250 µm was faster than that of 500 µm. This is because the smaller the particle size the larger the surface area available for drug release. The drug release from formulation containing Eudragit RS only was slow, but when RL was used in combination with RS, stavudine release from micro spheres was faster that is: FA4 > FA1, FA5 >FA2 and FA6 >FA3 (see Fig. 3). This is due to the fact that the amount of quaternary ammonium groups of Eudragit RS is lower than that of Eudragit RL, which renders Eudragit RS is less permeable. It was also found that as the proportion of Eudragit RS was increased relative to drug concentration, the release rate of stavudine was decreased due to slower rate of diffusion through the polymer matrix. Release Kinetics The release mechanism of stavudine from various formulations was determined by comparing their respective correlation co-efficient (see Table 2.). It would appear that the mechanism of drug release from microspheres was diffusion-controlled. When the release rate constants of stavudine microspheres were compared, it was found to follow the following order: FA4 >FA1>FA5>FA6 >FA2 > FA3. CONCLUSION Stavudine microspheres was prepared successfully using the solvent evaporation method. Polymer: drug ratio, stirring speed and the content of magnesium stearate influenced the sphericity of the microspheres. The yield and entrapment efficiency were high for all formulations. It was observed that with increase in polymer concentration, the mean particle size of the microspheres increased but increasing the stirring speed and magnesium stearate content, resulted in a decrease in the mean particle size of microspheres. The assessment of the release kinetics revealed that drug release from stavudine microspheres followed Higuchi Model. It was suggested that mechanism of drug release from microspheres was diffusion-controlled. Controlled release without initial peak level achieved with these formulations may reduce dose frequency and side effects as well as improve patient compliance. ACKNOWLEDGEMENTS The authors greatly acknowledge Hetero Labs Ltd, Hyderabad, India, for supply of stavudine as a gift. The authors are grateful to Indian Institute of Technology (IIT), Kharagpur, India, University Science of Instrumental Center (USIC), Jadavpur, Kolkata, India and Indian Institute of Chemical Biology (IICB), Kolkata, India, for help in performing characterization studies. References
Copyright @2002-2006. TJPR Faculty of Pharmacy, University of Benin, Benin City, Nigeria The following images related to this document are available:Photo images[pr05007f3.jpg] [pr05007t1.jpg] [pr05007f2.jpg] [pr05007f1.jpg] |
| |||||||||


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}